Biochimica del sangue
Biochimica del sangue
Questo sito utilizza cookie, anche di terze parti. Se vuoi saperne di più leggi la nostra Cookie Policy. Scorrendo questa pagina o cliccando qualunque suo elemento acconsenti all’uso dei cookie.I testi seguenti sono di proprietà dei rispettivi autori che ringraziamo per l'opportunità che ci danno di far conoscere gratuitamente a studenti , docenti e agli utenti del web i loro testi per sole finalità illustrative didattiche e scientifiche.
Le informazioni di medicina e salute contenute nel sito sono di natura generale ed a scopo puramente divulgativo e per questo motivo non possono sostituire in alcun caso il consiglio di un medico (ovvero un soggetto abilitato legalmente alla professione).
Biochimica del sangue
Il plasma costituisce il 55% circa del volume ematico.
La percentuale del volume ematico occupata dalle cell ematiche è chiamata valore ematocrito.
Il siero si ottiene lasciando coagulare il sangue o il plasma: si forma una massa gelatinosa che a poco a poco si comprime spremendo fuori il siero.
Il siero ematico si differenzia dal plasma per la mancanza di fattori della coagulazione, essenzialmente fibrinogeno. Il sangue è reso incoagulabile per aggiunta di chelanti del Ca²+ (ossalato, citrato) o di eparina.
Il plasma
È costituito da:
- 90% di acqua
- 10% di soluti, fra cui
- le proteine (6,3-7,8g/100mL)
- sali inorganici (1,0-1,2 g/100mL)
- composti organici di varia natura (1-1,5 g/100mL)
Proteine plasmatiche
Il plasma contiene più di un centinaio di proteine (proteine semplici, glicoproteine, lipoproteine).
A scopo diagnostico esse vengono separate e determinate mediante elettroforesi su lastrine di gel di acetato di cellulosa o di agarosio, oppure su colonne capillari, a pH 8,6.
Le principali funzioni di tali proteine sono:
- il trasporto di molecole e ioni
- la funzione immunitaria
- antiinfiammatoria
- enzimatica
- ormonale
- altre funzioni particolari
Proteine di trasporto
Prealbumina
Un tetramero costituito da 4 identiche subunità e funge primariamente da proteina di trasporto per la tiroxina.
Albumina
La più abbondante proteina del plasma; è una proteina semplice di forma ellissoidale, poco viscosa, formata da un’unica catena polipeptidica contenente 9 domini, 17 ponti disolfuro e 5-6 tasche idrofobiche.
È biosintetizzata dal fegato nella forma di pre-albumina: dapprima avviene il distacco di un peptide-segnale N-terminale a livello del RER e poi quello di un esapeptide sempre a livello del terminale aminico, lungo la via secretoria verso il sangue.
È la principale responsabile della pressione oncotica del plasma (pressione osmotica esercitata dalle proteine del plasma à ~ 25mmHg) e quindi del riassorbimento di acqua a livello del capillare venoso.
Questa proprietà ha un ruolo fondamentale nel controllo degli scambi idrici fra capillari e liquido interstiziale; è per questa ragione che quando la concentrazione di albumina nel plasma diminuisce si ha edema, cioè accumulo di acqua negli spazi interstiziali.
L’abbondanza delle cariche negative superficiali abilita l’albumina a legare (reversibilmente) ioni (Ca²+, Ni²+, Zn²+) mentre la presenza delle tasche idrofobiche le consente di ancorare labilmente molecole idrofobiche fisiologiche (acidi grassi, bilirubina, triptofano, ormoni steroidei) o esogene, quali taluni farmaci (es. aspirina) per poi rilasciarle perifericamente.
Da qui il suo ruolo di trasportatore!
Globuline
Tra le globuline con funzione di trasporto abbiamo:
- la proteine di trasporto del retinolo (RBP)
- la globulina di trasporto del cortisolo (CBG) o trancortina
- la proteina legante gli ormoni sessuali (SHBG)
- le transcobalamine che trasportano la vitamina B12
- la transferrina, β-globulina glicosilata, che trasporta il ferro assorbito con la dieta o dismesso dalla milza, verso le sedi di utilizzo quali il fegato e il midollo osseo
- le aptoglobine formate da 2 paia di catene (α2 e β2) unite da ponti disolfuro, che legano in rapporti di 1:1 dimeri αβ di ossiemoglobina i quali possono ritrovarsi nel plasma a seguito di eventi emolitici intravasali
- l’emopexina (Hpx), β-globulina che si combina con l’eme rilasciato da emoglobina libera nel sangue
- la ceruloplasmina contenente 8 siti di legame per il rame che possiede di per sé attività enzimatica di tipo ossidasico, facilita l’assorbimento gastrointestinale del ferro e svolge un ruolo primario nel metabolismo del rame costituendone una riserva.
- le lipoproteine (chilomicroni, VLDL, IDL, LDL, HDL) addette al trasporto ematico di trigliceridi, colesterolo esterificato e non esterificato.
Proteine implicate nei fenomeno immunitari
Immunoglobuline (γ-globuline)
Prodotte dai linfociti B su stimolo di specifici antigeni, nell’ambito del meccanismo della reazione immunitaria a insulti solitamente esterni.
Sistema del complemento
Una ventina di proteine, indicate con le sigle C (da C1 a C9), B, D e P, termolabili oltre i 56°C.
Presenti allo stato inattivo vengono attivate per proteolisi da parte di vari stimoli; per es. in presenza di complessi antigene-anticorpo vi si aggregano, attivandosi ed esercitano su di essi azione proteolitica alla quale fa seguito la completa rimozione dei complessi stessi.
Analogamente si associano a superfici esogene (cell. batteriche), si attivano e innescano una cascata di eventi proteolitici che porta alla formazione di “pori” nella membrana causando lisi osmotica delle cell.
I frammenti proteolitici possono anche avere effetti funzionalmente rilevanti quali la stimolazione della fagocitosi e la promozione della chemotassi.
Proteine implicate nei processi infiammatori
Appartiene a questo gruppo la proteina C reattiva (CRP) prodotta dal fegato su stimolo dell’interleuchina-6.
La CRP, principale fattore di crescita per le plasmacell, aumenta di concentrazione nel corso di processi di infiammazione, di cui costituisce un indice importante, anche se aspecifico.
Proteine ad attività enzimatica
Si possono suddividere in 3 gruppi:
- enzimi la cui azione si svolge primariamente nel sangue, quali gli enzimi della coagulazione, della fibrinolisi, della regolazione della pressione (sistema renina-angiotensina); sono sintetizzati in altri organi e secreti nel sangue nella forma di pro-enzimi inattivi oppure di enzimi attivi.
- enzimi secreti dalle ghiandole esocrine (pancreas, prostata, mucosa gastrica ed intestinale) di solito presenti in quantità molto piccole.
- enzimi tissutali normalmente secreti nel sangue (es. la colinesterasi di origine epatica) o derivati dal disfacimento postnecrotico o apoptotico fisiologico di varie cell.
Merita una menzione particolare, il lisozima, costituito da un’unica catena di 129 aminoacidi secreto dalle ghiandole salivari e lacrimali nonché dai leucociti neutrofili; catalizza l’idrolisi del legame β-glicosidico fra il C1 dell’acido N-acetilmuramico e l’N-acetilglucosamina presente nei polisaccaridi della capsula batterica: da qui la sua azione battericida.
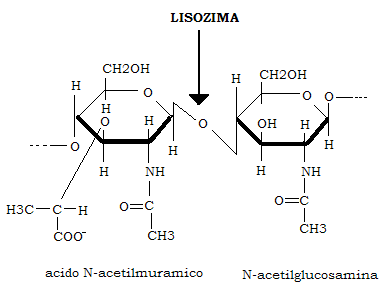
Molti di questi enzimi hanno un importantissimo significato diagnostico potendo il loro contenuto serico aumentare anche di decine o centinaia di volte a seguito di affezioni patologiche di organi o tessuti.
Quando l’aumento è responsabile di una specifica isoforma tipica di un organo o tessuto il riscontro assume il significato, estremamente rilevante, di “indicatore di lesione organo-specifica”.
Proteine con funzioni particolari
Crioglobuline
Un insieme eterogeneo di proteine plasmatiche che precipitano a temperatura fra 0 e 22°C e si ridissolvono a 37°C.
Comprendono immunoglobuline, componenti del fibrinogeno, fibronectina ecc..
Si classificano in 3 sottogruppi:
- di tipo I à costituito da singole immunoglobuline monoclonali
- di tipo II e III à costituite da 2 immunoglobuline di cui una policlonale e l’altra monoclonale, con associati componenti di altra natura
Le crioglobuline aumentano nel plasma in molti situazioni patologiche di tipo tumorale, infettivo, degenerativo o essenziale.
α1-Antitripsina
Inibisce le proteasi e agisce prevalentemente a livello polmonare.
α2-Macroglobulina
Inibisce varie proteasi presenti in piccole concentrazioni nel plasma.
β2-Microglobulina
Proviene dai linfociti e può essere escreta nelle urine. Aumenta di concentrazione nel plasma nell’insufficienza renale, nel mieloma multiplo ed in alcune neoplasie e ne è aumentata l’escrezione renale nell’insufficienza renale e in alcune tubulopatie congenite.
Orosumucoide
Chiamata anche α1-glicoproteina acida, è la proteina plasmatica più ricca di carboidrati; è sintetizzata dal fegato e costituita da 181 aminoacidi.
La sua concentrazione nel plasma aumenta in molte forme infiammatorie, in vari tumori e nella gravidanza.
α-Fetoproteina
Proteina simile all’albumina, contenuta ad elevata concentrazione nel plasma fetale; la sua concentrazione diminuisce drasticamente alla nascita.
Dal punto di vista diagnostico riveste duplice importanza:
- nella spina bifida e in altre malformazioni della colonna vertebrale del feto fuoriesce nel liquido amniotico; il suo riscontro in questo liquido e a volte anche nel sangue materno consente la diagnosi prenatale di spina bifida
- un aumento di questa proteina nell’adulto è diagnostica di epatoma primario (tumore maligno del fegato) e possibilmente di altre malattie tumorali del fegato.
Biosintesi e turnover delle proteine plasmatiche
Le proteine del plasma sono sintetizzate nel fegato; la sintesi dell’albumina e del fibrinogeno è particolarmente rapida.
La sede epatica della sintesi delle proteine plasmatiche spiega la diminuzione del livello plasmatico di albumina e di fibrinogeno nella cirrosi epatica o in seguito ad epatectomia (resezione segmentale del fegato).
L’albumina plasmatica costituisce una importante riserva di aminoacidi per l’organismo.
Il breve turnover delle proteine plasmatiche (in media 10 giorni di emivita) implica un’elevata velocità di degradazione!
Elettroliti plasmatici
La concentrazione salina del plasma è, nel suo insieme, molto simile a quella dell’acqua marina; tale somiglianza costituisce il fondamento principale della ipotesi dell’origine della vita nell’acqua del mare.
Gli ioni prevalenti nel plasma e nei liquidi extracell sono i Na+ ed i Cl¯.
Nel liquido intracell i cationi prevalenti sono i K+.
Questa differente concentrazione tra Na+ e K+ fuori e dentro gli eritrociti è dovuta all’attività della “pompa Na+/K+”.
Sodio ioni
La concentrazione plasmatica dei Na+ e la pressione osmotica dei liquidi extracell, di cui i Na+ sono i principali determinanti, aumenta nelle seguenti condizioni:
- malattie renali, che inducono ritenzione di Na+
- insufficiente perfusione renale, implicante una diminuita filtrazione glomerulare dei Na+
In entrambe queste condizioni oltre ai Na+ aumentano nel plasma anche i Cl¯.
- iperaldosteronemia, che aumenta il riassorbimento tubulare dei Na+ e nel contempo diminuisce il riassorbimento dei K+, incrementandone la perdita.
Potassio ioni
Modificazioni plasmatiche degli ioni K+ incidono più profondamente che non quelle dei Na+ sulla funzionalità delle cell, specialmente quelle eccitabili, quali le cell cardiache.
- Ipokaliemia, una diminuzione plasmatica dei K+ si può verificare per perdita renale di questi ioni, che si verifica nelle seguenti condizioni: acidosi, eccesso di Na+, disidratazione, ipercatabolismo proteico e iperaldosteronemia. La ipokaliemia produce alterazioni della eccitabilità a livello cardiaco, che si manifestano con aritmia, tachicardia e ipotensione.
- Iperkaliemia, si può verificare nella insufficienza renale acuta ed in condizioni come l’acidosi, che determinano fuoriuscita di K+ dalle cell.
L’aumentata concentrazione plasmatica di K+ determina aritmia cardiaca, bradicardia, fibrillazione ventricolare ed arresto cardiaco in diastole, quando la concentrazione dei K+ supera i 10 mEq/L.
Altri componenti del plasma
Oltre all’acqua, alle proteine e ai minerali, il plasma (o siero) contiene metaboliti di diversa origine tissutale o d’organo, oppure di origine esogena attraverso l’alimentazione.
Ne sono un esempio il glucosio, l’urea, l’acido urico, l’acido lattico, i corpi chetonici ecc..
L’efficacia dei vari sistemi di regolazione fan sì che la concentrazione di alcuni di questi componenti del plasma (o siero) rifletta la condizione di omeostasi dell’intero organismo.
Le cellule del sangue
Tutte le cell ematiche originano da un progenitore comune, l’emocitoblasto (cellula staminale emopoietica), a seguito di un processo di differenziamento in senso eritropoietico (eritrociti), leucopoietico (granulociti o leucociti polimorfonucleati) e trombopoietico (megacariociti, progenitori delle piastrine).
Nelle fasi iniziali del processo di differenziamento le cell staminali emopoietiche si distinguono in:
- linfoidi che evolvono in linfociti T e B
- mieloidi che danno origine agli eritrociti, ai granulociti, ai monociti e ai megacariociti da cui le piastrine.
Globuli rossi o eritrociti
Sono cell prive di nucleo e degli altri organelli cell, fra cui i mitocondri. Alla formazione dell’ATP nei globuli rossi non contribuisce quindi la fosforilazione ossidativa mitocondriale.
Sono dotati di una vita media di 120 giorni, hanno forma a disco biconcavo con diametro di 6,7-7,7 µm e spessore di 1,7-2,5 µm.
Nel loro interno i costituenti citoplasmatici sono in soluzione e quindi liberamente diffusibili all’esterno, allorché la membrana eritrocitaria venga strutturalmente alterata o per immersione dell’eritrocita in un mezzo ipotonico o per azione di agenti tensioattivi.
L’emoglobina è presente all’interno del globulo rosso alla concentrazione di 7mM, pari al limite della sua solubilità.
Membrana eritrocitaria
Una delle più importanti proprietà fisiche dei globuli rossi è la deformabilità che consente loro di passare lungo capillari aventi un diametro inferiore.
La deformabilità è a sua volta espressione della plasticità dell’eritrocita.
Quando per alterazioni della membrana questa capacità di deformazione viene meno, gli eritrociti rimangono impigliati nelle maglie dei tessuti (specie milza) e vengono distrutti.
La elasticità e altre proprietà dipendono fondamentalmente dalle proteine che la costituiscono, in prevalenza glicoproteine; fra questa la glicoforina, costituita da 131 residui di aminoacidi, con l’estremità N-terminale protrudente dalla superficie esterna e quella C-terminale rivolta al citosol.
L’estremità citosolica è costituita da residui di aminoacidi polari (aspartato e glutammato).
L’estremità esterna lega, tramite residui di serina o treonina, 14 catene tetrasaccaridiche e, tramite un residuo di asparagina, un singolo, più complesso, oligosaccaride arboriforme.
Le glicoforina costituisce il 5% in peso degli eritrociti umani ed ogni eritrocita contiene 500mila molecole di glicoforina.
Un’altra importante proteina è la banda 3 o proteina trasportatrice degli anioni (scambia con l’ambiente extraeritrocitario l’HCO3¯ con il Cl¯), costituita da 2 monomeri giustapposti, con una principale porzione idrofobica che attraversa parecchie volte lo spessore della membrana e presenta i terminali sia aminico sia carbossilico sul versante citosolico della membrana stessa.
Altra proteina è la spettrina, proteina estrinseca sottesa alla superficie citosolica della membrana eritrocitaria a guisa di un reticolo tubulare ancorato da un lato a proteine intrinseche della membrana, dall’altro a proteine filamentose citosoliche che insieme compongono il citoscheletro eritrocitario.
La spettrina è suscettibile di fosforilazione reversibile e di conseguenza è in grado di assumere rapporti modulabili con le proteine intrinseche della membrana e con quelle citoplasmatiche.
Il citoscheletro eritrocitario comprende l’anchirina che fa da ponte tra la banda 3 e la spettrina e la banda 1 che, insieme all’actina, stabilisce i punti di connessione fra le strutture fibrillari della spettrina.
Altre importanti proteine della membrana eritrocitaria, anche se presenti in più piccole quantità sono:
- il trasportatore del glucosio (GLUT3), capace di trasportare 180 molecole di glucosio al secondo
- il trasportatore dei nucleosidi
- il trasportatore degli aminoacidi
- i trasportatori di ioni, ATP-dipendenti: Na+, K+-ATPasi e Na+, H+-ATPasi.
Metabolismo degli eritrociti
Glicolisi: formazione dell’ATP e sua utilizzazione
I globuli rossi ricavano tutto l’ATP di cui necessitano dalla glicolisi, processo che fornisce anche il 2,3-difosfoglicerato necessario per il rilascio dell’O2 dall’emoglobina.
Il lattato che si forma effluisce dai globuli rossi e viene usato soprattutto dal fegato.
Gli equivalenti riducenti, in forma di NADPH(H+), necessari per il mantenimento del ferro emoglobinico allo stato ferroso e del glutatione allo stato ridotto, vengono invece forniti dal ciclo dei pentoso-fosfati.
Il glucosio costituisce quindi il substrato unico e indispensabile per tutte le funzioni energetiche dell’eritrocita!!
L’ATP che si forma viene interamente usato per il funzionamento della “pompa Na+/K+”, l’ATPasi di membrana che promuove il trasporto contro gradiente dei Na+ verso l’esterno e dei K+ verso l’interno; detto scambio crea un potenziale negativo all’interno della cell: per questo il trasporto mediato dalla ATPasi è “elettrogenico”.
Formazione del 2,3-bifosfoglicerato
L’altro prodotto correlato alla glicolisi, importante per la funzionalità del globulo rosso è il 2,3-bifosfoglicerato (BPG).
Nell’ambito della molecola dell’emoglobina la molecola del BPG forma dei legami crociati salini fra le cariche negative dei 2 gruppi fosforici e del gruppo carbossilico e le cariche positive di residui di istidina e lisina delle due subunità β.
Questa interazione tra emoglobina e BPG facilita il rilascio dell’O2 dalla ossiemoglobina ai tessuti.
Il BPG riveste notevole importanza anche nel sangue usato per le trasfusioni.
Negli eritrociti conservati in un medium di glucosio e citrato, la concentrazione di BPG diminuisce in pochi giorni fino ad un decimo del valore originale.
A causa della elevata affinità che conseguentemente l’emoglobina acquista per l’O2, i globuli rossi trasfusi sono pressoché inefficienti a rilasciare ossigeno ai tessuti!
D’altro canto i globuli rossi non sono in grado di incorporare BPG dall’esterno. Si può tuttavia ripristinare BPG nel globulo rosso aggiungendo al medium inosina, capace, a differenza del BPG, di penetrare entro il globulo rosso.
Nell’eritrocita la molecola di ribosio, che si libera dalla inosina, può essere metabolizzata nel ciclo dei pentoso-fosfati e successiva glicolisi e produrre BPG.
Ciclo dei pentoso-fosfati: funzione del NADPH(H+) e del glutatione ridotto
Il NADPH(H+) prodotto nel ciclo dei pentoso-fosfati viene utilizzato per la riduzione del glutatione eritrocitario.
Il mantenimento del glutatione allo stato ridotto (GSH) è una condizione importante per la stabilità del globulo rosso; infatti la ossigenazione della emoglobina ad ossiemoglobina è un processo a rischio, in quanto una piccola aliquota di O2, anziché legarsi alla emoglobina per formare ossiemoglobina, sottrae un elettrone alla emoglobina per formare O2¯˙ e metaemoglobina (MetHb):
Hb (Fe²+) + O2 à MetHb (Fe³+) + O2¯˙
MetHb (Fe³+) à La emoglobina aggrega e precipita nei cosiddetti corpi di Heinz!
Nelle 24 ore questo processo produce in ogni globulo rosso circa 107 molecole di O2¯˙; queste molecole di superossido (ROS), se non tempestivamente rimosse, risulterebbero altamente lesive per la stabilità della membrana eritrocitaria!! (Provocano invecchiamento cell, neoplasie e malattie cardiovascolari)
La detossificazione dei ROS (specie reattive dell’ossigeno) è un’attività significativa degli eritrociti!!
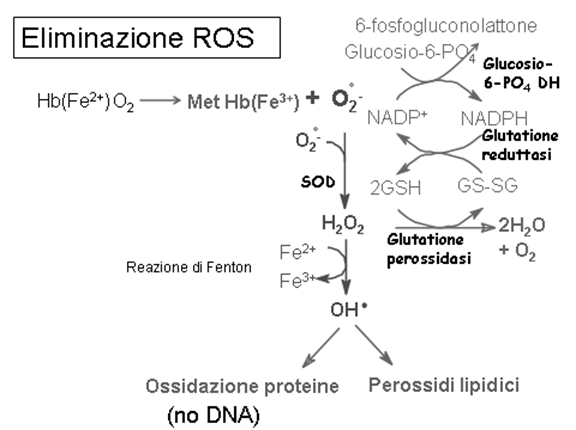
SOD = superossido dismutasi
La SOD può essere:
- mitocondriale, Mn²+ dipendente
- citosolica, Cu²+ o Zn²+ dipendente
GSH = glutatione ridotto
GSSG = glutatione ossidato
O2˙ ¯ à superossido
H2O2 à perossido di idrogeno
OH ˙ à idrossile
La glutatione perossidasi contiene selenio ed è presente in tutti i tessuti ma particolarmente abbondante a livello dei globuli rossi. La glutatione reduttasi ripristina il glutatione.
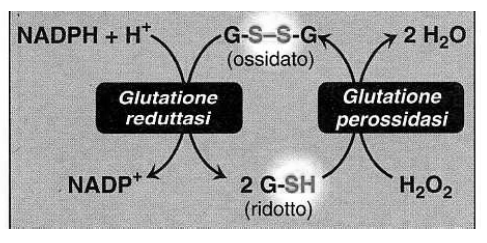
Il NADPH(H+) proviene principalmente dalla via dei pentosi (glucosio-6-fosfato deH e 6-fosfogluconato deH).
La reazione di Fenton è l’acquisizione non enzimatica di e¯ dal Fe²+ e si basa sull’interazione in ambiente acquoso tra acqua ossigenata e ioni di ferro ferroso.
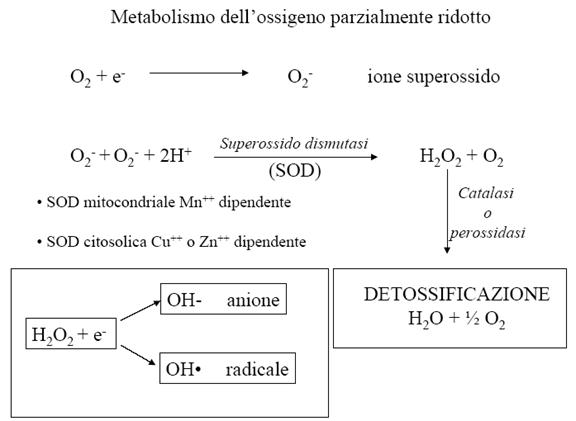
Nella reazione catalizzata dalla SOD, una molecola di superossido funge da ossidante e l’altra da riducente.
I ROS sono potenti ossidanti per la presenza di elettroni spaiati negli orbitali esterni (sottraggono elettroni ad altre molecole per completare il loro ottetto), sono quindi radicali liberi. Si formano sia nei globuli rossi ([O2] elevata) che nella maggior parte della altre cell.
Le molecole più vulnerabili all’azione dei radicali liberi sono i lipidi in quanto le catene carboniose lineari degli acidi grassi sono praticamente “allo scoperto”.
Le proteine grazie alla loro conformazione spaziale riescono invece a proteggersi meglio dall’azione dei ROS.
Gli acidi nucleici per il fatto di essere presenti prevalentemente nel nucleo sono raggiungibili con più difficoltà.
Numerose patologie collegate alle ROS derivano da un alterato equilibrio tra produzione e rimozione di ROS, condizione nota come “stress ossidativo”.
Sistemi antiossidanti à - superossido dismutasi
- catalasi
- glutatione perossidasi
Sistemi di assorbimento disponibili à - albumina
- bilirubina
- acido urico
- glutatione
Vitamine ad azione antiossidante à - Vit. A
- Vit. C
- Vit. E
Esistono 3 modalità di formazione di ROS:
- Reattiva, correlata cioè alle difese dell’organismo; le ROS vengono sintetizzate in cell fagocitarie quali i neutrofili e i macrofagi per uccidere i batteri da essi fagocitati; gli enzimi coinvolti sono
- NADPH ossidasi (in entrambi i tipi di cell)
- mieloperossidasi (solo nei neutrofili), una eme proteina
- Ipossica, ovvero la transitoria carenza di ossigeno a livello generale o in una regione specifica del corpo porta alla formazione di radicali liberi
- Energetica (metabolica), correlata alla catena respiratoria mitocondriale (cattivo funzionamento degli elementi della catena di trasporto elettronico) e quindi dipendente dall’apporto calorico (sovraccarico energetico, dieta ipercalorica)
I gruppi sanguigni
La superficie degli eritrociti è rivestita da un complesso mosaico di determinanti antigenici, costituiti da glicoproteine e glicolipidi di membrana, la cui specificità di gruppo è tuttavia impartita dalla porzione glucidica.
Nell’uomo sono stati identificati 15 determinanti antigenici, dei quali i gruppi A, B, 0 e Rh sono clinicamente importanti.
La presenza nel siero dei corrispondenti anticorpi (agglutinine) determina l’agglutinazione degli eritrociti.
Le piastrine e la coagulazione del sangue
Funzione delle piastrine
Fondamentali per il processo di coagulazione, le piastrine o trombociti sono prodotti dalla disintegrazione esplosiva dei megacariociti.
La loro vita media è di 9-12 giorni e hanno un elevatissimo contenuto idrico (86-88% del peso secco); per il resto sono costituite, in ordine, da proteine, lipidi e glicidi.
Sono dotate di un robusto citoscheletro con fasci sottoplasmatici e microtubuli connessi con proteine contrattili quali l’actina e la miosina, le quali costituiscono, rispettivamente, il 10-20% e il 15-20% delle proteine totali.
Caratteristiche delle piastrine sono pure i granuli di secrezione, che possono contenere varie sostanze fra cui fibrinogeno, trombospondina, alcuni fattori della coagulazione, nucleotidi, istamina, serotonina, adrenalina, basi puriniche.
Il sistema tubulare denso, derivante dal RE, contiene Ca²+, acido arachidonico ed enzimi quali la fosfolipasi A2 e la cicloossigenasi: in questo compartimento avviene la sintesi di trombossani e di altri eicosanoidi.
Contiguo a questi organuli è il sistema canalicolare costituito da invaginazioni della membrana plasmatica.
Attivazione, adesione e aggregazione delle piastrine
Una lesione del letto vascolare è responsabile di 2 eventi:
1) l’esposizione del collagene presente nella matrice extracell agli elementi figurati del sangue, in particolare alle piastrine
2) l’innesco del processo di attivazione della protrombina in trombina
La superficie delle piastrine contiene recettori sia per il collagene (complesso glicoproteico GPIa-IIa detto anche α2 β1-integrina) sia per la trombina (complesso glicoproteico GPV).
Il complesso collagene/GPIa-IIa è stabilizzato dall’interazione tra fattore di von Willebrand (fvW) e un altro specifico recettore piastrinico, il complesso glicoproteico GPIb-V/IX.
Le combinazioni dei complessi GPIa-IIa e GPIb-V/IX rispettivamente con il collagene e il fvW sono responsabili della parziale attivazione di una fosfolipasi A2 presente nel citosol e della adesione al subendotelio delle piastrine.
Le piastrine cambiano di forma, diffondono nel subendotelio e si predispongono alla aggregazione in situ.
Il recettore della trombina, interagendo con il suo ligando, si attiva e innesca il processo trasduzionale di segnali che porta all’attivazione della fosfolipasi di membrana C-β (PLC-β) con formazione di diacilglicerolo e inositolo-1,4,5-trifosfato (IP3).
In presenza di diacilglicerolo e piccole quantità di Ca²+, viene attivata la PKC che catalizza la fosforilazione della proteina pleckstrina (detta anche proteina P47) che a sua volta provoca la liberazione del contenuto dei granuli di secrezione, tra cui ADP, dando inizio alla aggregazione piastrinica.
L’IP3 provoca l’apertura dei canali del Ca²+ del sistema dei tubuli densi e la fuoriuscita nel citosol dello ione. Si raggiunge una concentrazione di Ca²+ sufficientemente alta per saturare la calmodulina e consentire la fosforilazione della cinasi delle catene leggere della miosina (calmodulina dipendente), la quale fosforilando la miosina promuove l’aggancio con l’actina.
L’aumento della concentrazione citosolica del Ca²+ porta alla completa attivazione della fosforilasi A2, che catalizza il distacco dai fosfolipidi di acido arachidonico, il quale risulta disponibile alla conversione in trombossano A2, ad opera della cicloossigenasi.
Questo eicosanoide fuoriesce dalla cell, e legando ad un proprio recettore, incentiva l’asse trasduzionale centrato sulla attivazione della PLC-β: pertanto sia la liberazione del Ca²+ nel citosol sia la secrezione di sostanze attive, tra cui l’ADP, ne risultano potenziati.
L’ADP secreto favorisce a sua volta il processo di aggregazione.
Il quadro complessivo della membrana delle piastrine attivate è favorevole alla esposizione di un complesso glicoproteico GPIIb che legandosi al fibrinogeno favorisce ulteriormente l’aggregazione piastrinica e la stabilizza nella forma di tappo piastrinico o trombo bianco.
Sostanze quali la serotonina e l’adrenalina, liberate per secrezione dai granuli piastrinici, e la vasopressina, esercitano un’azione agonistica sulla aggregazione piastrinica!!
Al contrario ligandi, quali la prostaciclina, che provocano la produzione di cAMP inibiscono (o riducono) l’aggregazione piastrinica in quanto il cAMP blocca la liberazione di Ca²+ dal sistema tubulare denso e inibisce sia la fosfolipasi A2 che la cicloossigenasi.
Endoperossidi, prostacicline, prostaglandine e trombossani nella aggregazione piastrinica
L’aspirina inibisce la trasformazione dell’acido arachidonico negli endoperossidi, potenti fattori aggreganti.
I trombossani che si formano nelle piastrine dagli endoperossidi, hanno un potere aggregante 5 volte superiore a quello degli stessi endoperossidi.
D’altro canto le prostacicline, che si formano dagli endoperossidi a livello delle pareti vasali integre, hanno invece azione antiaggregante e neutralizzano, in condizioni di integrità dell’endotelio vasale, l’azione dei trombossani.
Se l’endotelio è invece alterato, la produzione in loco della prostacicline e delle prostaglandine, che hanno pure azione antiaggregante, è deficitaria e prevale l’azione aggregante dei trombossani.
Formazione del coagulo permanente
L’aggregazione piastrinica contribuisce alla formazione del coagulo provvisorio. Per la formazione del coagulo permanente è necessaria la fibrina, presente nel sangue sotto forma di precursore solubile, il fibrinogeno.
Gli eventi che portano alla formazione del coagulo permanente sono di tue tipi:
- estrinseco, indotto da fattori non presenti nel sangue, ma che si liberano o si rendono disponibili a seguito di lesioni vasali (tissutali)
- intrinseco, sostenuto da fattori tutti presenti nel sangue.
I due processi hanno in comune le due tappe terminali della coagulazione:
- conversione protrombina à trombina
- conversione fibrinogeno à fibrina
Conversione del fibrinogeno in fibrina
Il fibrinogeno è una glicoproteina presente nel sangue nella concentrazione di 3-4 g/L circa.
La molecola è costituita da 3 paia di catene: 2α, 2β e 2γ, tenute insieme da legami disolfuro.
Il fibrinogeno viene sintetizzato nel fegato e, immesso nel sangue, ha un’emivita di 4 giorni.
Per azione della trombina, che catalizza l’idrolisi del legame Arg-Gly presente in ciascuna delle 2 catene α e β, il fibrinogeno (solubile, detto anche fattore I della coagulazione) viene trasformato in fibrina (insolubile, detta anche fattore Ia).
Contemporaneamente si liberano i fibrino peptidi A e B, costituiti l’uno da 16 e l’altro da 14 aminoacidi, dall’estremità N-terminale di ciascuna catena α e β.
La fibrina neoformata è in forma di monomeri, che si associano testa a coda e lato a lato per formare un precipitato insolubile denominato coagulo molle; questo per azione del fattore XIIIa, che crea legami covalenti tra le estremità dei monomeri di fibrina, viene stabilizzato in coagulo duro.
I legami covalenti creati dal fattore XIIIa, dotato di attività transglutaminasica, sono legami glutamil-lisinici, formatisi per interazione di un residuo di glutammina ed uno di lisina con eliminazione di una molecola di ammoniaca.
Il fattore XIIIa si forma per attivazione del fattore XIII, una proteina presente nel plasma e nelle piastrine, per l’azione successiva della trombina e dei Ca²+.
Durante il passaggio da coagulo molle a coagulo duro nella rete del polimero di fibrina vengono intrappolate piastrine prima ed eritrociti poi con formazione del trombo bianco e trombo rosso, rispettivamente.
Conversione della protrombina in trombina
La trombina, formata da 2 catene polipeptidiche è detta anche fattore IIa ed è presente nel sangue in forma di un precursore inattivo: la protrombina o fattore II.
La conversione della protrombina in trombina richiede l’azione catalitica del fattore Xa, una endopeptidasi detta anche fattore di Stuart.
L’azione di questo fattore richiede l’intervento dei Ca²+, di fosfolipidi di provenienza piastrinica e di un altro fattore proteico, la proaccelerina o fattore V, nella sua forma attiva fattore Va, che si può considerare un fattore accessorio del fattore X.
Dal punto di vista strutturale la trasformazione fattore II à IIa implica il distacco di un frammento peptidico N-terminale e la formazione di una asola tenuta chiusa da un legame disolfuro.
Nel plasma sono presenti 4 inibitori naturali della trombina dei quali il più rilevante è l’antitrombina III, seguita dall’α2-macroglobulina e da 2 proteasi tripsinosimili poco rilevanti.
Il processo intrinseco
Il contatto con il collagene della parete vasale alterata induce l’attivazione del fattore di Hageman (fattore XII di natura proteica) in XIIa. La causa di questa attivazione è una modificazione conformazionale della molecola proteica indotta dal contatto con cariche negative della superficie, che la rende suscettibile all’azione di varie proteasi.
Il fattore di Hageman attivato (XIIa) con la sua attività protesica dà inizio alla cascata di eventi.
Esso induce per proteolisi l’attivazione del fattore XI in XIa; questo a sua volta attiva il fattore di Christmas (fattore IX) per proteolisi Ca²+-dipendente.
Il fattore di Christmas può essere attivato anche dalla proconvertina attivata (fattore VIIa) in presenza di Ca²+ e di fosfolipidi acidici.
La tappa successiva consiste nell’attivazione del fattore di Stuart (fattore X) da parte del fattore IXa in presenza di Ca²+, fosfolipidi e fattore antiemofilico VIIIc. Infine il fattore IXa catalizza la trasformazione della protrombina in trombina.
Il processo estrinseco
Nel processo estrinseco l’attivazione del fattore di Stuart avviene con un altro meccanismo, nel quale intervengono la proconvertina attivata (fattore VIIa) ed un fattore tissutale detto tromboplastina.
La tromboplastine è una proteina transmembrana costituita da 263 aminoacidi di cui 219 sono esposti verso l’esterno. È questa la porzione della tromboplastina interessata alla formazione del complesso macromolecolare che dà inizio al processo estrinseco.
I fattori II, V, VII, IX e X sono sintetizzati nel fegato e la loro sintesi richiede, fra l’altro, l’intervento di una carbossilasi che, introducendo un carbossile in alcuni residui glutamminici, crea altrettanti residui γ-carbossilglutammilici (questi residui legano elettrostaticamente i Ca²+ necessari per l’attivazione dei predetti fattori).
Fibrinolisi del coagulo
Pochi giorni dopo la sua formazione il coagulo viene dissolto per l’azione proteolitica della plasmina, serina-proteasi, che si forma da un precursore denominato plasminogeno, sintetizzato nei reni.
Il plasminogeno viene attivato a plasmina per rottura di un singolo legame peptidico e formazione di 2 catene polipeptidiche tenute insieme da un ponte disolfuro; tale rottura è catalizzata da alcuni enzimi quali la urochinasi e la streptochinasi.
Urochinasi e streptochinasi reagiscono con il plasminogeno anche in assenza di coagulo; per contro l’endotelio vascolare elabora un “attivatore tissutale del plasminogeno” (t-PA) che attiva il plasminogeno solo in presenza del coagulo.
Per questa ragione il t-PA può considerarsi l’attivatore fisiologico del plasminogeno!!
Emoglobina
L’emoglobina ha una forma quasi sferica con un diametro di circa 5,5nm; è una proteina tetramerica contenente 4 gruppi prostetici eme, uno per ciascuna subunità.
L’emoglobina A (dell’adulto) contiene 2 tipi di globine e cioè 2 catene α e 2 β.
La struttura quaternaria dell’emoglobina è caratterizzata da interazioni molto forti tra le 4 subunità.
L’emoglobina esiste in 2 conformazioni principali:
- lo stato T (bassa affinità)
- lo stato R (alta affinità)
L’emoglobina può legare ossigeno in entrambi gli stati, ma l’affinità dello stato R per il ligando è molto più elevata.
Il legame dell’ossigeno alla proteina stabilizza lo stato R.
Quando l’ossigeno non è disponibile, lo stato T è più stabile e quindi questa è la conformazione predominante della deossiemoglobina.
Il legame dell’ossigeno ad una subunità dell’emoglobina nello stato T innesca una modificazione conformazionale che la converte nello stato R.
Tale legame è di tipo cooperativo e la curva che lo rappresenta è di tipo sigmoide (a forma di S) ad indicare che dopo che si è legato l’O2 alla prima subunità, la transizione TàR rende più facile il legame di una seconda molecola di O2.
Oltre a trasportare l’ossigeno, l’emoglobina trasporta anche 2 prodotti finali della respirazione cell: H+ e CO2 dai tessuti ai polmoni e ai reni dove sono escreti.
La CO2 viene idratata in forma di bicarbonato:
CO2 + H20 à H+ + HC03¯
Questa reazione è catalizzata dalla anidrasi carbonica, un enzima particolarmente abbondante negli eritrociti.
L’idratazione della CO2 determina un aumento della concentrazione di H+ (diminuzione pH) nei tessuti.
Il legame dell’ossigeno all’Hb è profondamente influenzato dal pH e dalla concentrazione di CO2!
L’H+ promuove infatti la dissociazione dell’O2 dall’emoglobina: è un effettore eterotropico negativo!
Alla diminuzione del pH è quindi associata la liberazione di O2 da parte dell’Hb à Effetto Bohr.
Anche la CO2 promuove la dissociazione dell’O2 dall’Hb, agendo attraverso la generazione di H+.
Alcune molecole di CO2 legano direttamente Hb nella forma di carbammato (-NHCOO¯) e causano la stabilizzazione dello stato T.
Il 2,3-bifosfoglicerato riduce profondamente l’affinità dell’emoglobina per l’ossigeno e quindi O2 e BPG hanno effetti allosterici opposti!
Esso si lega all’emoglobina nella cavità tra le subunità β nello stato T; questa cavità è rivestita da aminoacidi con gruppi R carichi positivamente che interagiscono con i gruppi carichi negativamente del BPG.
Al contrario dell’O2, una sola molecola di BPG si lega ad ogni tetramero di Hb; agisce abbassando l’affinità dell’Hb per l’O2 e stabilizzando lo stato T.
In assenza di BPG l’emoglobina viene convertita più facilmente nello stato R.
Il BPG è il principale responsabile della “cooperatività”.
L’altitudine aumenta la sintesi di BPG, incrementando la cooperatività e spostando la curva sigmoide ulteriormente verso destra.
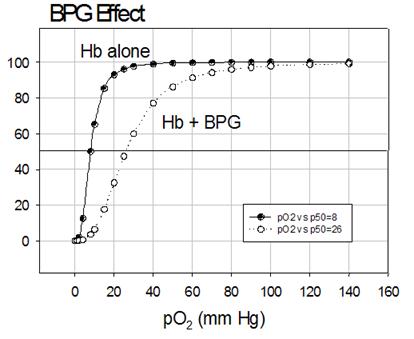
A basse concentrazioni di pO2 la via più rapida per adattarsi è l’incremento della sintesi di BPG, più efficiente dell’incremento della sintesi di Hb.
L’emoglobina fetale (Hbf)
L’emoglobina fetale è predominante nella vita fetale e nella prima infanzia; nei bambini e negli adulti se ne trovano solo tracce.
Il dosaggio negli adulti è eseguito per confermare la diagnosi in caso di certe forme di microcitemia (presenza nel sangue di microciti, ovvero di eritrociti con dimensioni inferiori alla norma).
L’emoglobina fetale lega più debolmente il BPG rispetto a quella dell’adulto e poiché il BPG abbassa l’affinità dell’Hb per l’O2, ne consegue che l’Hbf ha un’affinità maggiore per l’O2 rispetto all’Hb A.
Fonte http://www.medwiki.it/sites/default/files/Biochimica%20del%20sangue.doc
Sito web da visitare: http://www.medwiki.it/
Autore del testo: non indicato nel documento di origine
Parola chiave google : Biochimica del sangue tipo file : doc
Visita la nostra pagina principale
Biochimica del sangue
Termini d' uso e privacy