DNA e RNA
DNA e RNA
Questo sito utilizza cookie, anche di terze parti. Se vuoi saperne di più leggi la nostra Cookie Policy. Scorrendo questa pagina o cliccando qualunque suo elemento acconsenti all’uso dei cookie.I testi seguenti sono di proprietà dei rispettivi autori che ringraziamo per l'opportunità che ci danno di far conoscere gratuitamente a studenti , docenti e agli utenti del web i loro testi per sole finalità illustrative didattiche e scientifiche.
Struttura e funzioni del DNA
Un percorso di ricerca
LE BASI MOLECOLARI DELL'EREDITARIETÀ |
|
1869 |
Miescher scopre il DNA nel nucleo cellulare; lo chiama "nucleina" |
1902 |
Sutton, analizzando mitosi e meiosi, scopre le basi cromosomiche dell'ereditarietà. |
1910 – 1939 |
Morgan, Muller e altri pongono le basi della teoria del gene: unità più piccola del cromosoma che trasporta le informazioni genetiche. Le mutazioni sono variazioni dei geni. |
1928 |
Griffith (microbiologo inglese), lavorando su pneumococchi virulenti e non scopre il "principio trasformante", una molecola in grado di trasferire informazione da un ceppo batterico a un altro (meccanismo oggi noto come "trasformazione batterica"). E' la conferma che i geni sono molecole o parti di molecole. |
Anni '30 |
I cromosomi sono formati sia da proteine che da DNA; quale molecola contiene l'informazione genica? La scelta degli studiosi cade sulle proteine, più variabili e ritenute più complesse., anche perché lunghe ricerche effettuate dal biochimico Levene hanno fatto ritenere erroneamente che il DNA fosse una piccola molecola identica in tutte le specie. |
1944 |
Avery, con una brillante serie di esperienze, dimostrò che il "principio trasformante" di Griffith era DNA. Ma le sue conclusioni non furono accettate e solo 9 anni dopo ci fu la dimostrazione conclusiva. |
Anni '40 |
Delbruck, Luria e altri lavorano sui virus in grado di infettare batteri, chiamati batteriofagi o fagi. Scoprono che tali virus iniettano nel batterio la molecola che contiene le informazioni genetiche. |
1952 |
Herschey e Chase, lavorando con fagi marcati con isotopi radioattivi del fosforo e dello zolfo, dimostrano definitivamente che è il DNA e non le proteine a portare i geni. |
LA STRUTTURA CHIMICA DEL DNA |
|
1869 |
Miescher scopre il DNA nel nucleo cellulare.l (vedi sopra) |
Anni '20 |
Levene formula l'ipotesi del tetranucleotide, secondo la quale il DNA è una piccola molecola senza variabilità. |
1938 |
Todd ed altri si resero conto che il DNA era una molecola ben più grande e complessa. I risultati furono pubblicati soltanto nel 1945. |
Dal 1930 al 1950 |
Bragg, Bernal, Astbury, poi Pauling, Perutz e altri, studiarono la struttura di molecole biologiche complesse, come le proteine, con il metodo della diffrazione a raggi X. |
1948 – 1952 |
Chargaff si rese conto che nel DNA la quantità di adenina e di timina erano uguali, come pure quelle di guanina e citosina. Ciò in tutte le specie. |
1951 – 1953 |
Fondamentali lavori di Wilkins e, soprattutto, di R. Franklin, sulla diffrazione dei raggi X da parte del DNA. |
1953 |
Watson e Crick, sfruttando anche i risultati ottenuti fino a quel momento, scoprono la struttura a doppia elica del DNA. |
COME SI ESPRIMONO I GENI |
|
Anni '40 |
Beadle e Tatum, studiando la muffa Neurospora crassa formulano l'ipotesi che il gene contenga le informazioni per un enzima specifico. Ciò permette alla cellula di svolgere una certa azione. "Un gene ® un enzima". |
1953 |
Meselson e Stahl scoprono la duplicazione semiconservativa del DNA |
1957 |
Crick formula il "dogma centrale della biologia": |
1961 |
Matthaei e Nirenberg, su un'idea del 1054 di Gamow, iniziano la decifrazione del codice genetico: ogni aminoacido è codificato da una tripletta di nucleotidi chiamata codone. |
1950 – 1960 |
Vengono chiariti i meccanismi della sintesi proteica: trascrizione e traduzione. |
1960 |
I francesi Jacob e Monod fanno i primi studi sulla regolazione genica: nei batteri scoprono un meccanismo di regolazione con repressori chiamato modello dell'operone. Successivamente scoprono altri meccanismi di regolazione genica negli eucarioti. |
Fonte: http://liceocorso.scuolaer.it/documenti/biologia/DNA%20scoperta.doc
Autore del testo: non indicato nel documento di origine
Dna Finger-Printing
Il Dna finger-printing è un metodo di identificazione che consiste nel comparare frammenti di acido deossiribonucleico (DNA) provenienti da diversi individui. Il Dna è il materiale genetico contenuto nel nucleo delle cellule di tutti gli esseri viventi. Nei mammiferi i filamenti di Dna sono raggruppati in strutture chiamate cromosomi. Con l'eccezione dei gemelli omozigoti identici, il DNA di ogni individuo è unico. La prima operazione da eseguire per la realizzazione del Dna finger-printing, è estrarre un campione di Dna da un tessuto o da un liquido del corpo, come ad esempio capelli, sangue o saliva. Il campione deve essere ora suddiviso utilizzando enzimi di restrizione, la cosiddetta digestione, i siti di restrizione sono i punti dove gli enzimi di restrizione vanno a tagliare, la differenza che si evidenzierà sarà la frammentazione: due Dna uguali produrranno lo stesso numero di frammenti con lo stesso peso molecolare.
I segmenti ottenuti sono organizzati in base alla loro grandezza usando:
- un processo chiamato elettroforesi;
- evidenziati con un colorante fluorescente;
- esposti per lo studio ai raggi ultra violetti.
Il DNA finger-printing è stato ultilizzato per la prima volta come tecnica di identificazione nel 1985.
Originariamente utilizzato per rilevare la presenza o meno di malattie genetiche, è in seguito risultato utile per le indagini criminali e nella medicina legale. Infatti se, analizzando campioni di materiale biologico provenienti, ad esempio, da un luogo particolare come lo scenario di un delitto otteniamo che il DNA finger-printing prodotto da due diversi campioni coincide, allora i due campioni prelevati provengono dalla stessa persona e quindi possiamo dedurre che il sospettato è colpevole.
La prima prova per una condanna criminale, basata su test del DNA negli Stati Uniti, si è presentata nel 1988. Nelle indagini criminali, le impronte digitali del DNA sono state derivate dalle prove raccolte sulla scena del crimine, e confrontate con le impronte digitali del DNA dei sospetti. La prova del DNA può condannare o meno un sospettato.
In generale le corti hanno accettato come prove affidabili i test sul DNA, tuttavia il DNA finger-printing è discutibile sotto alcuni punti di vista, come ad esempio l'esattezza dei risultati, il costo del test e il possibile abuso di questa tecnica.
L'esattezza del DNA finger-printing è stata messa in discussione per parecchi motivi. In primo luogo perchè il DNA viene suddiviso in strisce che possono non essere uniche per ciascun individuo; ricerche su grande scala hanno messo in evidenza che test sull'unicità del DNA non sono state effettuate. In più questi test vengono condotti in laboratori privati che non sempre possono seguire le procedure standard e controlli di qualità. Inoltre dato che l'uomo deve in seguito interpretare il test, vi è anche il problema dell'errore umano.
Il DNA finger-printing è un test costoso e alcune persone con pochi capitali potrebbero non essere in grado di difendersi adeguatamente da accuse che si basano sul test in questione. L'uso molto diffuso di questa tecnica a scopo di identificazione potrebbe portare alla creazione di un database contenente tutte le nostre impronte di DNA. Il database potrebbe potenzialmente essere utilizzati per scopi illeciti come identificare individui con malattie che potrebbero portare a discriminazioni sul luogo di lavoro.
Enzimi di Restrizione
La parola "enzima" viene dal greco enzyme (letteralmente "nel levito"), e venne usata per la prima volta nel 1878 dal biochimico Kuhne per contrapporre i "fermenti organizzati", cioè i microorganismi interi, ai fermenti presenti in estratti di microorganismi, in particolare nel lievito di birra. Dal suo contesto iniziale, l’uso del termine enzima si è poi allargato in quanto tutti gli organismi viventi, dai batteri più primitivi all’uomo, producono enzimi necessari per la vita.
Dal punto di vista funzionale, gli enzimi sono proteine che svolgono il ruolo di catalizzatori. I catalizzatori infatti rendono più efficienti, o addirittura possibili, determinate reazioni chimiche, che nel caso degli enzimi avvengono all’interno della cellula; senza gli enzimi le medesime reazioni procederebbero troppo lentamente o potrebbero non avvenire affatto. Il potere catalizzante deriva dalla loro struttura molecolare: gli enzimi avvicinano, legandoli, i reagenti altrimenti dispersi in una soluzione, li dispongono nella posizione ottimale affinché reagiscano efficacemente e soprattutto stabilizzano i prodotti intermedi della reazione. In questa maniera le specie chimiche reagiscono in un microambiente, detto sito attivo, che facilita la rottura di alcuni legami chimici e la creazione di nuovi, con la conseguente formazione di nuove molecole.
L’enzima di restrizione è un particolare tipo di enzima che è in grado di riconoscere specifiche sequenze di basi lungo il filamento di Dna, e di "tagliare" esattamente in corrispondenza di queste sequenze.
La sua scoperta avvenne casualmente tramite un’osservazione: quando in un ceppo del batterio Escherichia Coli si introduceva del DNA proveniente da un ceppo diverso, questo DNA veniva letteralmente tagliato a pezzetti. Fu postulato che vi fossero degli enzimi prodotti dai batteri responsabili di questa attività. In seguito fu dimostrato che lo stesso accadeva quando i batteri venivano infettati da un virus: il DNA del virus veniva immancabilmente ridotto in frammenti inattivi, cioè la crescita del virus si disse ristretta in un determinato ceppo di batteri. Di qui il nome di enzimi "di restrizione". Gli enzimi di restrizione sono ritenuti essere una sorta di sistema immunitario primitivo dei batteri. Come fu successivamente scoperto essi attaccano solamente il DNA esterno, ad esempio di un virus, perché il DNA dei batteri presenta delle modificazioni chimiche, per la precisione l’aggiunta del gruppo metile (CH3), che lo rendono inattaccabile dai propri enzimi. Non passò molto tempo e il primo di questi enzimi venne scoperto e purificato in batteri della specie Haemophilus influenzae e quindi battezzato HindII. Attualmente il loro numero ha superato abbondantemente il migliaio e sono venduti da industrie biotecnologiche che devono il loro successo al grande uso che si fa di essi nella ricerca.
La caratteristica che rende così preziosi gli enzimi per lo studio del DNA è che ognuno di essi riconosce, lega e taglia il DNA in una sequenza ben precisa. Ad esempio, EcoRI (isolato da Escherichia coli RY13) riconosce la sequenza GAATTC, mentre BamHI (isolato da Bacillus amyloliquefaciens H) la sequenza GGATCC.
Sebbene le sequenze riconosciute siano alle volte simili, ogni enzima è in grado di discriminarle con estrema precisione. In questa maniera è possibile tagliare l’intero genoma umano in frammenti più maneggiabili, detti frammenti di restrizione; o, conoscendone la sequenza, ritagliarne fuori un certo gene; o ancora, modificare un gene accorciandolo; operazioni queste che preludono a qualunque studio di biologia molecolare.
Mediante l’azione di un enzima detto DNA ligasi, i frammenti possono essere poi saldati ad un altro frammento di DNA di qualunque natura, purchè trattato con lo stesso enzima di restrizione (ciò da origine a estremità complementari, che possono poi unirsi).
La possibilità di costruire in vitro molecole ibride di DNA è stata determinata dalla scoperta di enzimi, le endonucleasi di restrizione, che tagliano la molecola di DNA a livello di siti specifici, dando così origine a frammenti particolari.
La maggior parte dei batteri produce uno o più enzimi di restrizione, probabilmente perché essi offrono il vantaggio selettivo di proteggere in qualche misura la cellula dall’infezione da parte dei fagi che contengono DNA a doppia elica.
Finora sono state caratterizzate oltre 2000 endonucleasi di restrizione differenti, che vengono identificate con una sigla di tre lettere: la prima, maiuscola, è la lettera iniziale del nome del Genere, e le altre due, minuscole, sono le prime due lettere della stessa specie batterica. Ad esempio, EcoR1 e HindIII sono endonucleasi di restrizione presenti rispettivamente in particolari ceppi di E. coli e di Haemophilus influenzae.
Elettroforesi
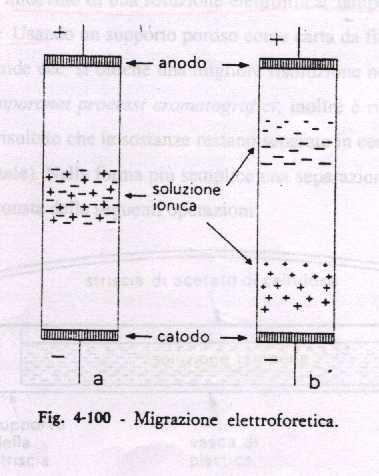
Esempio di migrazione elettroforetica di una soluzione ionica.
Le particelle cariche positivamente migrano verso il polo negativo e viceversa.
A seconda delle dimensioni le particelle si dispongono in zone diverse del gel:
le più grandi più vicine ai pozzetti di caricamento e le più piccole più avanti.
L'elettroforesi è un metodo di separazione basato sulla diversa velocità di migrazione di particelle elettricamente cariche attraverso una soluzione, sotto l'influenza di un campo elettrico.
Il principio base è quello di un setaccio molecolare attraverso il quale le diverse molecole vengono fatte passare: la velocità di migrazione dipende dalla massa, dalla dimensione, dalla carica e dalla forma delle varie particelle, ossia dalla loro mobilità elettroforetica. Questa grandezza è il rapporto tra la velocità della particella (cm/s) e il campo elettrico utilizzato (Volt/cm). La mobilità elettroforetica, essendo una funzione del rapporto tra carica e raggio, è diversa da una particella ad un'altra, applicando un campo elettrico ad una miscela ionica le varie specie migreranno con velocità diversa a seconda delle rispettiva mobilità.
L'elettroforesi è un metodo di separazione eccellente per macromolecole ed in particolare per proteine e frammenti di DNA; la sua semplicità e la sua velocità rendono tale sistema il più diffuso ed utilizzato.
La migrazione elettroforetica che avviene su un supporto solido di natura porosa imbevuto di una soluzione elettrolitica (tampone che permette il passaggio della corrente) prende il nome di elettroforesi zonale in quanto le sostanze restano separate in zone ben distinte.
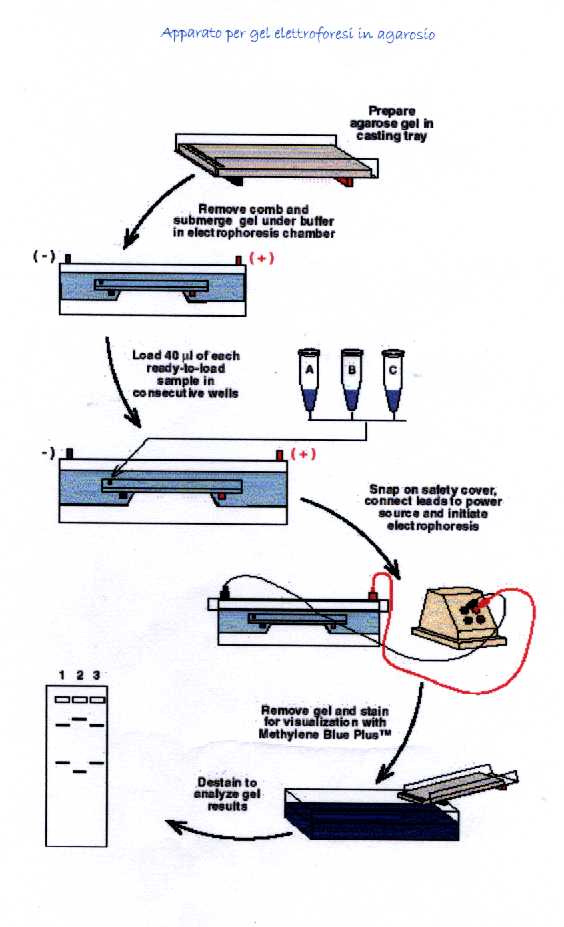
Gel elettroforesi di DNA
L'elettroforesi su gel separa i frammenti di DNA in base alla loro dimensione: frammenti di lunghezza minore sono meno ostacolati nella loro corsa attraverso le maglie del gel, quindi si troveranno nella parte bassa di quest'ultimo, mentre i frammenti più grossi si troveranno localizzati nella parte prossima ai pozzetti di caricamento.
Grazie alla presenza dei gruppi fosfato (PO43-) il DNA è carico negativamente e migrerà quindi verso il polo positivo.
La maggior parte delle separazioni elettroforetiche di campioni di DNA viene eseguita su gel di agarosio, in quanto esso ha pori di dimensioni adeguate. I campioni di DNA vengono preparati aggiungendo un colorante (come il blu di bromofenolo) e inoltre una volta terminata la separazione, anche il gel viene colorato (solitamente viene utilizzato il bromuro di etidio, un colorante fluorescente che viene stimolato utilizzando la luce ultravioletta di un transilluminatore).

Cella elettroforetica
Fattori che influenzano la separazione elettroforetica:
- tampone e suo pH:
il tampone ha una duplice funzione:
1. rende possibile il passaggio della corrente
2. mantiene costante il pH durante il processo elettroforetico
Il tampone più usato è di EDTA (Etilen diammina [contiene due gruppi amminici NH2] tetracetico acido)
- differenza di potenziale ed intensità di corrente:
Un incremento di d.d.p. permette di ridurre notevolmente i tempi di esecuzione della corsa ma causa anche un aumento del calore prodotto, da tenere sotto controllo in quanto potrebbe denaturare alcune sostanze.
- diffusione:
Il processo elettroforetico s'accompagna sempre a fenomeni di diffusione. Questa non incide sulla mobilità ma sulla larghezza della bande, comportando un effetto negativo ai fini della separazione.
Fonte: http://www.liceogioia.it/EspDidattiche/DNAfingerprinting.doc
Autore del testo: non indicato nel documento di origine
DNA e RNA
ESTRAZIONE DEL DNA
Per effettuare analisi e studi genetici è necessario poter accedere al DNA dell’organismo che si intende analizzare. Nell’uomo è possibile estrarre il DNA da cellule di diverso tipo. Il DNA è infatti identico in ogni cellula dell’organismo. I diversi protocolli di estrazione, quindi, si differenziano per il metodo applicato ma non per il risultato finale.
I tessuti preferenziali da cui si estrae il DNA sono: il sangue (dai globuli bianchi), l’epitelio buccale, i capelli. Le tecniche di prelievo delle cellule da questi tessuti sono tipicamente scarsamente invasive.
I protocolli di estrazione sono stati messi a punto proprio con l’obiettivo di ottenere la maggior quantità di DNA con il minimo disturbo del soggetto da campionare.
Ecco i punti cruciali dell’estrazione del DNA:
- assegnazione etichetta
- lisi cellulare
- estrazione del DNA
- purificazione del DNA dalle proteine residue
- raccolta e conservazione del DNA.
1. ASSEGNAZIONE ETICHETTA
Il campionamento, inteso come raccolta del materiale biologico da cui estrarre il DNA, deve essere strettamente coordinato alla fase di estrazione. Infatti è necessario etichettare in modo univoco il campione biologico prelevato, in modo che la stessa etichetta identifichi il DNA che si estrae da esso. Abitualmente, per etichettare i campioni, si utilizza un codice composto da lettere e numeri. Le lettere esprimono sinteticamente caratteristiche come il luogo di origine del campione o il motivo per cui è stato campionato. La numerazione, solitamente, è progressiva e cronologicamente correlata ai diversi momenti di campionamento.
2. LISI CELLULARE
La lisi comprende la rottura delle cellule e il rilascio del DNA in esse contenuto. Si utilizzano abitualmente tamponi contenenti SDS e proteinasi K (digerisce le proteine della membrana plasmatica e gli istoni), con aggiunta di agenti denaturati per le proteine come la guanidina idrocloride.
3. ESTRAZIONE
L’estrazione del DNA avviene in seguito alla dissociazione dei complessi DNA-PROTEINE tramite denaturazione e/o proteolisi. In particolare, si può procedere in 2 modi:
1) aggiunta di cloroformio o fenolo alla fase acquosa del lisato.
Questa azione provoca la denaturazione delle proteine.
2) legame del DNA presente nella soluzione acquosa ad una resina di silice. In questo secondo caso l’estrazione e la purificazione del DNA avvengono in un unico step.
4. PURIFICAZIONE DEL DNA DALLE PROTEINE RESIDUE
Questa fase consente di separare il DNA dalle altre macromolecole presenti in soluzione. Solitamente si procede precipitando il DNA in presenza di isopropanolo.
5. RACCOLTA E CONSERVAZIONE DEL DNA
La fase finale della procedura di estrazione comporta la raccolta del DNA (sotto forma di eluato o di precipitato). Il DNA è conservato risospeso in un tampone e congelato a -20°C. Prima di procedere alla fase di conservazione è necessario valutare la qualità/quantità di DNA ottenuto. A tal fine è possibile stimare la concentrazione del DNA mediante lettura spettrofotometrica, o tramite visualizzazione su gel d’agarosio e confronto con DNA a concentrazione nota.
Fonte: http://biologia.unical.it/openlab/documenti/protocolli/IL_NOSTRO_DNA_preliminari.doc
Autore del testo: non indicato nel documento di origine
GLI ACIDI NUCLEICI
Tutti gli organismi contengono acidi nucleici sotto forma di acido deossiribonucleico (DNA) e ribonucleico (RNA)
Il DNA è il depositario dell’informazione genetica che viene trascritta (cioè copiata) in molecole di RNA. L’RNA contiene il codice per sintetizzare specifiche proteine.
Una molecola acido nucleico è un polimero costituito da monomeri detti nucleotidi. Ciascun nucleotide è costituito da tre molecole:
- uno zucchero pentoso
- una base azotata
- una molecola di acido fosforico
Uno zucchero pentoso è uno zucchero la cui molecola è costituita da 5 atomi di carbonio. La base azotata è legata al carbonio 1 dello zucchero pentoso, mentre l’acido fosforico (H3PO4) è legato al carbonio 5 che è esterno all’anello dello zucchero.
I nucleotidi si legano tra loro medianti legami fosfodiestere che collegano il C3 del pentoso di un nucleotide al C5 del nucleotide successivo. In questo modo l’acido fosforico impiega due dei suoi tre gruppi acidi nel legame fosfodiestere 3-5. il gruppo acido restante conferisce alla molecola di acido nucleico particolari caratteristiche:
- proprietà acide
- capacità di legarsi con proteine basiche (istoni)
- basofilia (la molecola può essere facilmente colorata con coloranti basici)
Lo zucchero pentoso è il ribosio nell’RNA e il deossiribosio nel DNA. La differenza tra i due è che al deossiribosio manca l’ossigeno legato al C2.
Le basi azotate si chiamano così perché contengono molti atomi di azoto e possono essere di due tipi: purine o pirimidine
I nucleotidi che costituiscono la molecola del DNA sono:
- adenosin monofosfato
- citidin monofosfato
- guanosin monofosfato
- timidin monofosfato
Quelli che costituiscono la molecola dell’RNA sono gli stessi, ad eccezione del timidin monofosfato che è sostituito dall’uridin monofosfato.
Un nucleoside è un nucleotide a cui manca il fosfato, cioè sono la combinazione di uno zucchero pentoso e di una base azotata. Per esempio, è un nucleoside l’adenosina:
Adenina → base azotata
Adenosina → nucleoside (adenina + zucchero pentoso)
Adenosin monofosfato → nucleotide (adenina + zucchero pentoso + fosfato)
L’adenosin trifosfato (ATP) è un particolare nucleotide con tre acidi fosforici, uniti tra loro con legami ad alto contenuto energetico. Infatti questa è una molecola fondamentale per la cellula perché rappresenta la principale forma di accumulo di energia.
La molecola di DNA è costituita da 2 catene polinucleotidiche le quali formano una doppia elica intorno allo stesso asse centrale. Esse sono antiparallele: i legami 3-5 fosfodiestere sono rivolti in direzione opposta. Questo vuol dire che se una catena inizia con il C3 libero e finisce con il C5 libero, l’altra catena è disposta in modo contrario.
Le due catene sono unite tra loro mediante legami a idrogeno che si instaurano tra le basi complementari. Le uniche coppie tra le quali è possibile il legame sono A-T e C-G.
Tra A e T si instaurano due legami a idrogeno mentre tra C e G se ne formano tre, quindi la coppia C-G è più stabile.
A=T C≡G
La sequenza assiale di basi lungo una catena può variare molto, ma la sequenza della catena corrispondente deve essere complementare alla prima.
Le due catene possono essere separate tra loro rompendo il legame a idrogeno tra le coppie di basi. Questo può essere fatto mediante riscaldamento o un pH alcalino (fusione o denaturazione). Dopo la denaturazione si può riottenere la conformazione a doppia elica lasciando raffreddare lentamente il DNA, in modo che le badi possano riappaiarsi (rinaturazione)
La struttura dell’RNA è simile a quella del DNA, tranne che per la presenza del ribosio invece del deossiribosio e dell’uracile invece della timina. Inoltre L’RNA è costituito da una catena singola.
Ma le molecole di RNA, avendo estese regioni complementari all’interno di una stessa catena, spesso si ripiegano e tra le basi della catena si instaurano legami a idrogeno, formando delle anse a forcina.
Ci sono tre tipi di RNA:
- RNA messaggero (mRNA): porta l’informazione genetica per la sequenza di amminoacidi
- RNA transfer (tRNA): identifica e trasporta gli amminoacidi ai ribosomi
- RNA ribosomico (rRNA): rappresenta il 50% della massa dei ribosomi
LA CROMATINA
Nelle cellule degli eucarioti il DNA non è libero ma associato a piccole proteine dette istoni, in un complesso chiamato cromatina. Esistono cinque tipi differenti di istoni, tutti di natura basica. Per questo motivo possono instaurare uno stretto legame con il DNA, di natura acida.
I quattro istoni principali, H2A, H2B, H3 e H4 hanno ciascuno una composizione simile anche nelle specie più diverse, mentre l’istone H1 è differente da specie a specie.
La cromatina può presentare vari livelli di organizzazione:
- Aspetto moniliforme: si presenta come un filo di perle, cioè come una serie di perle spesse 10 nm e collegate tra loro da un filamento di DNA. L’aspetto moniliforme non rispecchia la vera struttura della cromatina, ma è un artefatto di tecnica, in cui viene eliminato l’istone H1. Si osserva quando la cromatina viene trattata per essere studiata al microscopio elettronico
- Fibra di 10 nm: con trattamenti meno drastici è stato osservato che la cromatina si presenta come una fibra di 10 nm in cui sono presenti delle unità ripetitive a stretto contato tra loro, dette nucleosomi. In ogni nucleosoma i quattro istoni H2A, H2B, H3 e H4 sono disposti in ottameri contenenti due molecole di ciascuna proteina attorno ai quali vi si avvolge il DNA, all’esterno. I nucleosomi sono a stretto contatto tra di loro e sono localizzati a intervalli di 200 coppie di basi di DNA. L’istone H1 si trova tra un nucleosoma e l’altro. Nella fibra di 10 nm la catena di DNA da 5 a 7 volte più compatta rispetto all’aspetto moniliforme, ma ancora 1000 volte meno rispetto ai cromosomi metafasici
- Fibra spessa: ha diametro variabile tra 20 e 30 nm e rappresenta la probabile struttura della fibra inattiva. Si forma in seguito all’avvolgimento della fibra di 10 nm in un solenoide. Il DNA raggiunge una compattezza pari a 40 volte quella iniziale, e per raggiungere la compattezza di un cromosoma metafasico deve ripiegarsi ancora un centinaio di volte
I CROMOSOMI
Nel corso della divisione cellulare la cromatina si condensa a formare i cromosomi, i quali sono circa 40.000 volte più densi della fibra di 10 nm. Essi sono strutture bastoncellari che servono a poter distribuire equamente il DNA tra le cellule figlie. I componenti dei cromosomi sono 4:
- CROMATIDI alla metafase ciascun cromosoma risulta formato da due strutture simmetriche, i cromatidi, contenenti ognuna un’unica molecola di DNA. Essi sono esattamente uguali (cromatidi fratelli) e sono uniti fra loro a livello del centromero
- CENTROMERO è la regione di attacco degli elementi del fuso mitotico sul cromosoma. È situato in un tratto più sottile del cromosoma detto costrizione primaria
- TELOMERO estremità dei cromosomi contenenti l’inizio e la fine della molecola di DNA che costituisce il cromatidio
- ORGANIZZATORE NUCLEOLARE si trovano in alcuni cromosomi che presentano costrizioni secondarie in cui ci sono regioni contenenti i geni che inducono la formazione del nucleolo
TEORIA UNINEMICA: ciascun cromatidio rappresenta un’unica molecola lineare di DNA con le proteine ad essa associate
I cromosomi vengono classificati in quattro tipi a seconda della forma determinata dalla posizione del centromero:
- METACENTRICI il centromero è posto a metà dei cromatidi e quindi le braccia sono uguali
- SUBMETACENTRICI le braccia sono di lunghezza differente
- ACROCENTRICI presentano un braccio cortissimo dove si trova la costrizione secondaria all’estremità della quale c’è una regione sferoidale detta satellite
- TELOCENTRICI il centromero è posto ad un estremo
La cromatina si condensa nel corso della mitosi e si decondensa alla telofase. Ma ci sono alcune regioni dei cromosomi che non si decondensano e rimangono compatte anche durante l’interfase. Queste regioni vengono chiamate eterocromatine, mentre le altre eucromatina.
Nell’eterocramatina il DNA è molto denso e si presenta in forma di fibra di 20-30 nm cioè si tratta di DNA inattivo.
L’esempio più noto è quello della coppia di cromosomi X nelle femmine dei mammiferi: uno dei due cromosomi è attivo e rimane eucromatico mentre l’altro è inattivo e costituisce il corpo di Barr nell’interfase.
Un altro esempio è quello del gatto di Spagna. Questo esemplare presenta un mantello a macchie nere e arancioni (a placche di tartaruga), ma solo le femmine. Questo perché la macchiatura un gene contenuto nel cromosoma X diventa eterocromatico e non attivo in alcuni gruppi di cellule e non in altri. In stadi precoci dell’embriogenesi, in ogni cellula della femmina di mammifero uno dei cromosomi X diventa inattivo a caso, di conseguenza nell’adulto si forma un mosaico in cui il 50% di cellule hanno un cromosoma X attivo di origine paterna e l’altro 50% un cromosoma X attivo di origine materna
IL CICLO CELLUARE
Una cellula in accrescimento presenta un ciclo cellulare che consiste essenzialmente in due periodi: l’interfase e la divisione. L’interfase presenta tre periodi che vengono chiamati fase G1,S e G2.
- Fase G1: la cellula raddoppia le sue dimensioni e aumentano di numero organuli e molecole organiche
- Fase S: avviene la duplicazione del DNA
- Fase G2: vengono prodotte le strutture necessarie alla divisione cellulare, la cromatina inizia a spiralizzarsi
- Mitosi: avvine la divisione cellulare
Duplicazione del DNA
La duplicazione del DNA avviene nella fase S (sintesi) del ciclo cellulare d’ogni cellula.
La replicazione del DNA è semi-conservativa: questo significa che ogni molecola figlia contiene una catena parentale e una neo sintetizzata.
Il primo passo verso la replicazione semi-conservativa è la separazione delle due catene complementari nel punto in cui deve cominciare la replicazione; in modo che ciascuna catena sia libera di fungere da stampo per la polimerizzazione di una nuova catena complementare. La polimerizzazione è catalizzata dall’enzima DNApolimerasi; che seleziona i deossinucleotidi trifosfati (d-ATP, d-TTP, d-GTP, d-CTP) e li lega uno dopo l’altro con legame fosfodiestere 3’-5’. L’enzima lega il fosfato presente su un deossinucleotide trifosfato al gruppo OH legato al carbonio 3’ del deossiribosio appartenente al nucleotide terminale della catena in crescita. Nella reazione i due gruppi fosfati terminali si staccano sotto forma di pirofosfato.
La DNApolimerasi catalizza solo l’allungamento unidirezionale di una catena, cioè può aggiungere nucleotidi solo all’estremità 3’ ma non all’estremità 5’. La DNApolimerasi può catalizzare l’aggiunta di nucleotidi all’estremità 3’ di una catena esistente ma non è in grado di iniziare una nuova catena. Per iniziarla è, infatti, necessario un innesco o primer cui aggiungere (all’estremità 3’) i nucleotidi in sequenza. Nella cellula fa da innesco una breve catena di RNA. L’innesco è sintetizzato come segue:
1. prima avviene una dissociazione circoscritta delle due catene su un tratto interno alla molecola di DNA
2. un enzima detto primasi catalizza la formazione di una breve catena di RNA a partire da ribonucleotidi trifosfati (ATP, UTP, CTP, GTP) [la primasi a differenza della DNApolimerasi è capace di dare inizio ad una catena]
3. l’innesco di RNA resta appaiato allo stampo, poi arriva la DNApolimerasi che estende la catena per aggiunta di deossinucleotidi all’innesco
4. naturalmente si forma un innesco su ciascuna catena parentale e i due inneschi hanno polarità opposta e quindi le due catene figlie crescono in direzioni opposte
5. man mano che le catene figlie si estendono alla loro estremità 3’ la doppia elica si apre a cerniera in entrambe le direzioni
6. il punto di rotolamento dell’elica si chiama forcella replicativi
7. man mano che la forcella avanza lascia dietro di se una zona a singolo filamento su ciascuna delle catene parentali. A questo punto si formano nuovi inneschi che sono poi prolungati per riempire a ritroso le parti non replicate delle catene parentali
8. gli inneschi sono poi rimossi quando i frammenti sintetizzati indietro (Okazaki) incontrano l’estremità 5’ dell’innesco del tratto precedente. Quando avviene l’incontro la DNApolimerasi stacca uno dopo l’altro i nucleotidi dell’innesco e aggiunge simultaneamente deossinucleotidi
9. quando tutto l’innesco è stato rimosso le due estremità sono unite dalla DNAligasi
Nella replicazione del DNA bisogna considerare il problema che la separazione delle due catene richiede la despiralizzazione della doppia elica. La doppia elica fa un giro completo ogni 10 paia di basi. Quindi ogni 10 basi separate e svolte a livello della forcella replicativa, la doppia elica a valle deve fare un giro in direzione opposta.
In E.coli la forcella si apre alla V=60000 paia di basi al minuto, il che richiederebbe che la doppia elica a valle facesse 6000 giri al minuto (negli eucarioti la V è 10 volte inferiore). La rotazione di tutto il DNA a valle della forcella è bloccato dalle numerose proteine associate perciò in mancanza di rotazione, la torsione della doppia elica a valle ostacolerebbe la despiralizzazione e impedirebbe la replicazione. Il problema è stato risolto grazie all’enzima topoisomerasi che crea rotture transitorie in un singolo filamento a breve distanza dalla forcella replicativa. Queste rotture si creano e si riparano rapidamente ad opera della stessa topoisomerasi.
http://riappunti.net/biologia/ACIDI%20NUCLEICI.doc
Autore del testo: non indicato nel documento di origine
IL MATERIALE GENETICO E’ IL DNA
Esperimento di Griffith
L'esperimento di Frederick Griffith del 1928 fu uno dei primi esperimenti a suggerire che i batteri sono in grado trasferire informazioni genetiche attraverso un processo noto come trasformazione.
In tal modo, esso aprì la strada alla determinazione di quale fosse la natura del materiale genetico.
Che esistesse una qualche sostanza in grado di trasmettere l'informazione genetica (il materiale genetico, appunto) era noto da tempo. Tra la fine del 1800 e i primi anni del 1900 venne proposto e dimostrato che il materiale genetico fosse racchiuso nei nuclei delle cellule (August Weismann) e in particolare nei cromosomi (teoria cromosomica dell'ereditarietà di Sutton, Boveri del 1902 dimostrata solo più tardi dagli esperimenti su Drosophila melanogaster di Thomas H. Morgan e dei suoi allievi).
Rimaneva però aperta la questione intorno alla materia costitutiva del materiale genetico.
L'ufficiale medico inglese F. Griffith in quegli anni studiava un batterio in grado di causare la polmonite: il pneumococco (Streptococcus pneumoniae). Nei suoi esperimenti fece uso di due ceppi batterici:
- Il ceppo S, detto anche liscio dal momento che produce colonie lisce e lucenti (grazie alla presenza di una capsula batterica polisaccaridica che avvolgeva ogni cellula). Questo ceppo è in grado di provocare la polmonite.
- Il ceppo R, detto anche rugoso dal momento che produce colonie dall'aspetto "rugoso" (a causa dell'assenza della capsula batterica). Questo ceppo non è in grado di provocare polmonite.
Schema dell'esperimento
Quando Griffith iniettò batteri R (ceppo IIR) in un topo, verificò che la cavia non si ammalava e non era possibile isolare questi batteri dai tessuti dell'animale.
- Quando il medico iniettò batteri S (ceppo IIIS) in topo, verificò che l'animale si ammalava, moriva ed era possibile isolare questi batteri dai tessuti dello stesso.
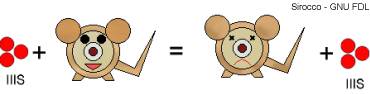
- Successivamente prese alcuni batteri IIIS e li uccise in seguito a shock termico. Iniettò poi questi batteri morti in un topo e come c'era d'aspettarsi il topo non si ammalò e non fu possibile isolare IIIS dai tessuti dell'animale.
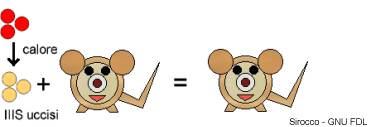
Da ciò si deduce che, per provocare la malattia, è necessaria la presenza della capsula e i batteri capsulati devono essere ovviamente vivi.
- A questo punto Griffith preparò una miscela in cui erano presenti batteri vivi IIR e batteri morti IIIS (uccisi col calore). Iniettò questa miscela in un topo: quello che ci si aspettava era la NON comparsa di malattia nell'animale (dal momento che non sarebbero dovute sussistere le condizioni appena citate). In realtà il topo si ammalò e morì; nei suoi tessuti si riscontrarono batteri IIIS vivi.
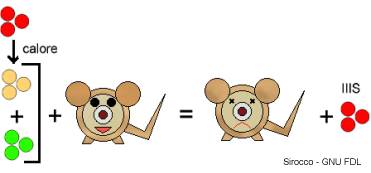
Conclusioni
Griffith propose l'unica spiegazione plausibile: alcuni batteri IIR, in seguito all'interazione con batteri morti IIIS si erano trasformati in IIIS.
Evidentemente all'interno dei IIIS morti doveva essere presente una qualche sostanza in grado di conferire ai batteri IIR che l'acquisivano la capacità di sintetizzare la capsula polisaccaridica.
Questa sostanza è il materiale genetico.
Griffith chiamò il materiale genetico principio trasformante. Erroneamente, come però la stragrande maggioranza degli scienziati suoi contemporanei, riteneva che questa sostanza dovesse essere di natura proteica.
A partire da questo importantissimo esperimento, Avery, MacLeod e McCarty nel 1943 dimostrarono che il materiale genetico in questione era il DNA (anche se la prova definitiva arrivò solo dagli esperimenti di Hershey e Chase del 1953
Esperimento di Avery, Mac Leod e McCarty
L'esperimento di Avery (Oswald Theodore Avery) e dei suoi colleghi Colin M. MacLeod e Maclyn McCarty risale al 1943 e rappresenta una delle esperienze fondamentali per l'avanzamento delle conoscenze nel campo della genetica e della biologia molecolare.
Tramite l'esperimento gli scienziati riuscirono a dimostrare che il cosiddetto principio trasformante (ovvero il portatore di informazioni geniche) scoperto nel 1928 da Griffith in seguito al suo famoso esperimento era il DNA.
Schema dell'esperimento di Avery
Avery si procurò una coltura di pneumococchi di tipo S. A questo punto lisò le cellule (cioè ne ruppe la parete e la membrana cellulare) in modo da ottenere una soluzione nella quale era disciolto il materiale contenuto nei batteri, il cosiddetto estratto cellulare o lisato cellulare.
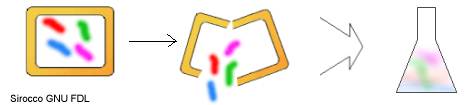
Il materiale genetico doveva presumibilmente essere uno dei diversi tipi di macromolecole biologiche presenti nei batteri: (proteine, polisaccaridi, acidi nucleici – ovvero DNA e RNA – e lipidi). Avery e colleghi riuscirono a separare l'estratto cellulare nelle varie componenti macromolecolari appena citate. Successivamente cercarono di capire quali di queste sostanze erano effettivamente in grado di trasformare batteri R avirulenti in batteri S virulenti. Le cavie sopravvivevano quando trattate con tutte le biomolecole tranne gli acidi nucleici: il materiale genetico doveva essere quindi DNA e/o RNA.
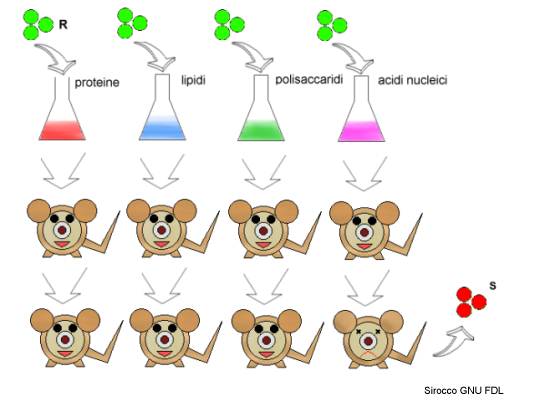
Per capire quale delle due sostanze fosse, divisero l'estratto contenente l'acido nucleico in due aliquote: una venne trattata con l'enzima ribonucleasi (RNasi) che degrada selettivamente l'RNA e non il DNA, l'altra venne invece trattata con deossiribonucleasi (DNasi) che degrada selettivamente il DNA e non l'RNA.
Ciò che si osservò era la trasformazione dei batteri R in batteri S solo in seguito all'aggiunta dell'aliquota trattata con RNasi. Il materiale genetico doveva allora essere necessariamente il DNA.
Le critiche all'esperimento
Il sopracitato esperimento non convinse tutti gli scienziati dell'epoca. Vi era infatti in quel periodo la convinzione diffusa (tra l'altro insita nello stesso Griffith) che il materiale genetico dovesse essere di natura proteica. Sia il DNA che le proteine sono dei polimeri. Nel caso delle proteine i monomeri (le unità base che ripetute danno il polimero) sono i 20 amminoacidi. Nel caso del DNA i monomeri sono solamente i 4 deossiribonucleotidi. Dal momento che l'informazione genetica doveva essere contenuta in queste macromolecole lineari, e considerata la grande differenza genetica tra le varie specie, pareva più sensato che il materiale genetico fosse natura proteica: in questo modo, rispetto agli acidi nucleici, sarebbero state possibili molte più combinazione tra i vari monomeri e di conseguenza l'informazione contenuta dalla macromolecola sarebbe stata maggiore.
Sotto la spinta di queste convinzioni, quello che veniva criticato ad Avery era la non completa purezza degli acidi nucleici utilizzati nell'esperimento: all'interno delle soluzioni contenenti DNA e RNA si ipotizzava fossero presenti anche tracce di proteine, le stesse proteine che gli scienziati scettici pensavano costituissero il materiale genetico.
In questo senso, pur non tralasciando l'importanza dell'evidenza sperimentale, non si può dire che l'esperimento di Avery, MacLeod e McCarty fosse la prova definitiva. Solo una decina di anni più tardi (1953) Hershey e Chase dimostrarono che il materiale genetico è costituito da DNA.
Esperimento di Hershey e Chase
L'esperimento di Alfred D. Hershey e Martha Chase prova definitivamente nel 1953 che il materiale genetico è costituito da DNA e non da proteine. In seguito a questi risultati incontrovertibili anche gli scienziati che avevano criticato l'esperimento di Avery, MacLeod e McCarty si convincono dell'importantissimo ruolo biologico del DNA.
Schema dell'esperimento
Hershey e Chase svolgevano studi su un fago, ovvero un virus in grado di infettare i batteri; in particolare il fago di loro interesse era noto come "T2". Questo virus è in grado di attaccare Escherichia coli (un batterio utilizzato spesso come modello in questo genere di studi).
All'epoca era noto che T2 era formato esclusivamente da DNA protetto da un involucro proteico.
I due scienziati prepararono in parallelo due colture di E. coli:
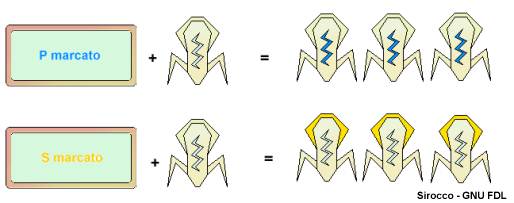
- Nel terreno di coltura della prima introdussero fosforo marcato radioattivamente (l'isotopo 32P).
- Nel terreno di coltura della seconda introdussero zolfo marcato radioattivamente (l'isotopo 35S).
I batteri delle due colture metabolizzarono da una parte il fosforo marcato e dall'altra lo zolfo marcato introducendo questi atomi radioattivi nelle biomolecole presenti all'interno delle cellule.
In particolare:
- Il fosforo marcato si troverà nei nucleotidi e di conseguenza anche negli acidi nucleici; non sarà presente invece in quantità significative nelle proteine.
- Lo zolfo marcato si troverà nelle proteine (in particolare nell'amminoacido cisteina) e non si troverà nei nucleotidi.
A questo punto i ricercatori infettarono parallelamente le due colture batteriche con il fago T2. Questo virus utilizza l'apparato biosintetico delle cellule di E. coli per replicare il proprio DNA e per sintetizzare le proteine del rivestimento e quindi per costruire virus completi che causeranno poi la lisi (rottura) della cellula batterica parassitata.
Dal momento che i nucleotidi e gli amminoacidi utilizzati nella sintesi del DNA e delle proteine virali sono quelli presenti all'interno della cellula batterica infettata (cresciuta nutrendosi degli isotopi radioattivi), ne risulta che i fagi sviluppati dall'infezione nella prima coltura avranno un DNA marcato radioattivamente, mentre quelli sviluppati dall'infezione della seconda coltura avranno il rivestimento proteico esterno marcato radioattivamente.
Hershey e Chase separarono i fagi neoformati (quelli marcati) dai due terreni di coltura e, separatamente, li utilizzarono per infettare altre due colture di E. coli, in questo caso cresciute su terreni "standard" privi di isotopi radioattivi.
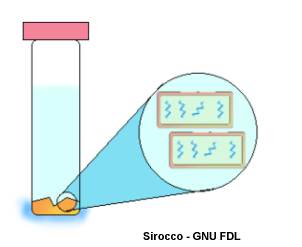
- Nel caso in cui i fagi infettanti avevano il DNA marcato, in seguito all'infezione gran parte della radioattività veniva misurata all'interno delle cellula batteriche colpite (e nel DNA di una parte dei nuovi fagi sviluppatisi in seguito a questa infezione).
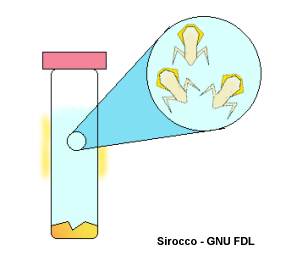
- Nel caso in cui i fagi infettanti avevano il rivestimento proteico marcato, la radioattività veniva misurata solamente all'esterno delle cellule batteriche colpite (e non era presente sul rivestimento proteico dei nuovi fagi sviluppatisi in seguito a questa infezione).
Il processo utilizzato per determinare se la radioattività provenisse dall'interno o dall'esterno delle cellule fu il seguente: dopo un certo tempo dall'inizio dell'infezione, il terreno di coltura veniva posto in un agitatore. La conseguente agitazione provocava il distacco del rivestimento proteico dei virus dalla membrana cellulare del batterio (in questo caso si parla di "ombre fagiche" poiché questi rivestimenti proteici non contengono il DNA che è già stato iniettato nella cellula). Il tutto veniva poi centrifugato: le cellule batteriche (contenenti eventualmente il DNA marcato) rimanevano sul fondo della provetta, mentre i rivestimenti proteici dei virus, distaccati dalle membrane cellulari dei batteri, rimanevano in sospensione. A seconda di dove si misurava la maggiore radioattività era possibile dedurre se la molecola marcata si trovasse o meno all'interno della cellula batterica.
Nel caso del DNA marcato la radioattività si misurò sul fondo della provetta (quindi all’interno dei batteri:
Nel caso delle proteine marcate la radioattività si misurò sul sopranatante (quindi all’esterno dei batteri):
Conclusione
Dal momento che il fago per replicarsi ha bisogno di introdurre all'interno della cellula ospite il suo materiale genetico per poter sfruttare l'apparato batterico di biosintesi, appare evidente che questo materiale genetico deve essere per forza il DNA poiché, come dimostrato, le proteine non entrano nella cellula colpita mentre il DNA sì.
A questo punto rimaneva aperta un'ultima questione: "Di cosa è fatto il materiale genetico degli "organismi" che non contengono DNA (i virus a RNA)?". La risposta a questa domanda non tardò ad arrivare in seguito a studi condotti sul virus del mosaico del tabacco (TMV) da Gierer e Schramm (1956) e da Fraenkel-Conrat e Singer (1957).
Argomenti tratti da Wikipedia, l'enciclopedia libera (con piccole semplificazioni e/o integrazioni).
Fonte: http://www.istitutovescovilenola.it/didattica/deangelis/appunti/biologia/IL%20MATERIALE%20GENETICO%20E%27%20IL%20DNA.doc
autore: non indicato nel documento di origine del testo
DNA e RNA
Visita la nostra pagina principale
DNA e RNA
Termini d' uso e privacy