Elettrochimica
Elettrochimica
ELETTROCHIMICA
Ossidazioni e riduzioni
Parlando dello ione H+ abbiamo visto che esso non può esistere da solo, allo stato libero, in quanto la sua carica elettrica e le sue ridotte dimensioni lo rendono estremamente reattivo. Esso pertanto può solo passare da un acido ad una base, durante una reazione chimica. L’elettrone possiede una carica elettrica dello stesso valore, anche se di segno opposto, di quella del protone, ma è circa 2.000 volte più piccolo. Esso dunque è ancor più reattivo del protone, pertanto non può esistere da solo, allo stato isolato, ma può solamente essere trasferito da una sostanza all’altra, durante una reazione chimica. Come per il protone, anche per il passaggio dell’elettrone è necessaria la presenza contemporanea di un donatore, che cede l’elettrone, e di un accettore, che lo acquista.
 Vediamo adesso quali sono le sostanze che cedono e quelle che accettano elettroni durante le reazioni chimiche. Gli elementi del I° gruppo, con la loro configurazione elettronica esterna nS1 debbono necessariamente cedere un elettrone per raggiungere la configurazione elettronica del gas nobile più vicino, mentre gli elementi del II° gruppo, con configurazione elettronica esterna nS2 debbono cederne due. Viceversa gli elementi del VII° gruppo, che hanno configurazione elettronica esterna nS2nP5, per raggiungere l’ottetto debbono acquistare un elettrone, mentre gli elementi del VI° gruppo, con configurazione elettronica esterna nS2nP4 debbono acquistarne due. E’ chiaro quindi che quando elementi dei primi gruppi incontrano elementi degli ultimi gruppi, i primi cedono elettroni ai secondi, come negli esempi che seguono:
Vediamo adesso quali sono le sostanze che cedono e quelle che accettano elettroni durante le reazioni chimiche. Gli elementi del I° gruppo, con la loro configurazione elettronica esterna nS1 debbono necessariamente cedere un elettrone per raggiungere la configurazione elettronica del gas nobile più vicino, mentre gli elementi del II° gruppo, con configurazione elettronica esterna nS2 debbono cederne due. Viceversa gli elementi del VII° gruppo, che hanno configurazione elettronica esterna nS2nP5, per raggiungere l’ottetto debbono acquistare un elettrone, mentre gli elementi del VI° gruppo, con configurazione elettronica esterna nS2nP4 debbono acquistarne due. E’ chiaro quindi che quando elementi dei primi gruppi incontrano elementi degli ultimi gruppi, i primi cedono elettroni ai secondi, come negli esempi che seguono:
Na + Cl → Na+ + Cl- → NaCl
Si può anche immaginare che questa reazione si costituita dalla somma di due semi reazioni che avvengono contemporaneamente:
Na → Na+ + e- Cl + e- → Cl-
 naturalmente l’elettrone perduto dal sodio è lo stesso che viene acquistato dal cloro. Sommando membro a membro le due semi reazioni si ottiene la reazione complessiva.
naturalmente l’elettrone perduto dal sodio è lo stesso che viene acquistato dal cloro. Sommando membro a membro le due semi reazioni si ottiene la reazione complessiva.
La reazione è esoergonica, se infatti soffiamo cloro elementare (che è un gas) su di un pezzo di sodio (che è un solido), la reazione avviene violentemente, con produzione di calore.
In verità il passaggio di elettroni può avvenire anche tra sostanze i cui atomi abbiano già raggiunto, almeno formalmente l’ottetto. Prendiamo come esempio la reazione tra idrogeno ed ossigeno: essa può avvenire, come nei razzi aerospaziali, attraverso una combustione, che produce calore, oppure può avvenire con produzione di energia elettrica, come nelle celle a combustibile. In entrambi i casi la reazione totale produce sempre lo stesso quantitativo di energia e può essere riassunta dalla seguente equazione: 2H2 + O2 → 2H2O.
Nell’idrogeno elementare il legame è omeopolare, gli elettroni sono quindi attratti in modo identico dai due nuclei d’idrogeno; nell’acqua invece vi è un legame eteropolare ed i due elettroni sono attratti maggiormente dal nucleo dell’ossigeno. In questo caso quindi il trasferimento degli elettroni è stato solo parziale. La reazione totale, bilanciata, può essere immaginata anche coma la somma delle seguenti semi reazioni:
4H → 4H+ + 4 e- 2O + 4 e- → 2O2- e quindi 4H+ + 2O2- → 2H2O.
In generale possiamo dire che:
- una sostanza che cede elettroni si ossida;
- una sostanza che acquista elettroni si riduce.
I due fenomeni debbono avvenire contemporaneamente, quindi la sostanza che cede elettroni riduce quella che li acquista, mentre la sostanza che acquista elettroni ossida quella che li cede. In base a questa ultima considerazione possiamo anche dire che:
- una sostanza che cede elettroni(e dunque si ossida) è un riducente;
- una sostanza che acquista elettroni(e dunque si riduce) è un ossidante.
Per complicare definitivamente le cose possiamo infine affermare che:
- un riducente si ossida, riducendo un ossidante;
- un ossidante si riduce, ossidando un riducente.
Le reazioni in cui vi è scambio di elettroni tra le sostanze che vi partecipano si dicono reazioni di ossidoriduzione o reazioni redox.
Il numero di ossidazione
Ma come possiamo capire se una reazione è una ossidoriduzione? E se lo è, come facciamo a capire quali sostanze si ossidano e quali si riducono? Allo scopo dobbiamo rifarci, ancora una volta, alla grandezza chimica chiamata numero di ossidazione. Infatti, contrariamente a quanto avviene nelle reazioni acido base, nelle reazioni di ossidoriduzione gli elementi coinvolti cambiano il loro numero di ossidazione.
 Rianalizziamo adesso i fenomeni dell’ossidazione e della riduzione alla luce del numero di ossidazione e ripartiamo dagli esempi già fatti in precedenza, calcolando, ad esempio, i numeri di ossidazione degli elementi nella reazione Na + Cl → Na+ + Cl-
Rianalizziamo adesso i fenomeni dell’ossidazione e della riduzione alla luce del numero di ossidazione e ripartiamo dagli esempi già fatti in precedenza, calcolando, ad esempio, i numeri di ossidazione degli elementi nella reazione Na + Cl → Na+ + Cl-

 Il sodio, che si ossida cedendo un elettrone, aumenta di una unità il suo numero di ossidazione; il cloro, che si riduce acquistando un elettrone, diminuisce di una unità il suo numero di ossidazione.
Il sodio, che si ossida cedendo un elettrone, aumenta di una unità il suo numero di ossidazione; il cloro, che si riduce acquistando un elettrone, diminuisce di una unità il suo numero di ossidazione.
Vediamo ora la reazione 2H2 + O2 → 2H2O.
In questo caso ogni atomo di idrogeno, che si ossida cedendo un elettrone, aumenta di una unità il proprio numero di ossidazione; l’atomo di ossigeno, che si riduce acquistando due elettroni, diminuisce di due unità il proprio numero di ossidazione.
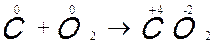 Prendendo sempre l’ossigeno come agente ossidante possiamo considerare anche la combustione del carbonio C + O2 → CO2
Prendendo sempre l’ossigeno come agente ossidante possiamo considerare anche la combustione del carbonio C + O2 → CO2
L’atomo di carbonio si ossida cedendo quattro elettroni ed aumenta di altrettante unità il proprio numero di ossidazione, viceversa ogni atomo di ossigeno si riduce, acquistando due elettroni e riducendo così di due unità il proprio numero di ossidazione.
Questi ultimi due esempi ci offrono l’opportunità di affermare che le reazioni di combustione sono reazioni redox nelle quali l’agente ossidante è l’ossigeno, detto anche comburente, mentre l’agente riducente è rappresentato da varie sostanze dette combustibili. Ecco che in parte abbiamo chiarito quali sono i rapporti tra ossidoriduzioni e produzione di energia.
Generalizzando quanto abbiamo ricavato dagli esempi sopra riportati, possiamo affermare che: un atomo che si ossida aumenta algebricamente il proprio numero di ossidazione di tante unità quanti sono gli elettroni che ha ceduto, un atomo che si riduce diminuisce algebricamente il proprio numero di ossidazione di tante unità quanti sono gli elettroni acquistati. Il numero di ossidazione, con le sue variazioni, diventa quindi l’elemento discriminante per distinguere le reazioni redox, dalle altre: infatti, come già detto, nelle reazioni di ossidoriduzione le sostanze che  reagiscono variano il loro numero di ossidazione, cosa che non fanno nelle reazioni di altro tipo, come le reazioni acido – base, di cui riportiamo un esempio:
reagiscono variano il loro numero di ossidazione, cosa che non fanno nelle reazioni di altro tipo, come le reazioni acido – base, di cui riportiamo un esempio:
Infine, all’interno di una reazione redox, il numero di ossidazione, con le sue variazioni algebriche, serve ad individuare chi si ossida, aumentando il proprio numero di ossidazione, e chi si riduce, diminuendo il proprio numero di ossidazione.
Le reazioni di ossidoriduzione
Ripetiamo adesso, riepilogando, quanto abbiamo già detto nei paragrafi precedenti. Nelle reazioni di ossidoriduzione gli elementi cambiano il loro numero di ossidazione perché si scambiano degli elettroni; gli elettroni non si trovano mai isolati, ma possono solo passare da un elemento all’altro: ci sarà dunque un elemento che cede elettroni ed un altro che li acquista. L’elemento che cede gli elettroni si ossida ed aumenta il proprio numero di ossidazione; viceversa l’elemento che acquista elettroni si riduce e riduce il proprio numero di ossidazione. Ossidazione e riduzione avvengono contemporaneamente, nel senso che un elemento si ossida solo se un altro si riduce e viceversa ed il numero di elettroni ceduti dagli atomi che si ossidano deve essere esattamente uguale al numero di elettroni acquistati dagli atomi che si riducono.
 Bilanciamento di una reazione di ossidoriduzione
Bilanciamento di una reazione di ossidoriduzione
Eccoci dunque all’argomento clou di ogni buon corso di chimica, quello sul quale ogni buon insegnante di chimica fa la sua selezione, trasmettendo così da una generazione di studenti all’altra l’idea della difficoltà della chimica.
Come avveniva nelle reazioni acido-base, bilanciare una reazione chimica significa scrivere degli opportuni coefficienti davanti alle sostanze reagenti ed a quelle prodotte, in modo tale che a sinistra ed a destra della reazione vi sia lo stesso numero di atomi di ciascun elemento. Non si debbono invece mai M variare i numeri in basso a destra degli elementi nei composti, in quanto si cambierebbe la composizione dei composti medesimi. Il bilanciamento di una reazione redox avviene attraverso una serie di passaggi che verranno di seguito elencati:
- calcolare il numero di ossidazione di tutti gli atomi presenti sia a destra che a sinistra;
- individuare gli elementi che si ossidano e quelli che si riducono;
- scrivere le semireazioni di ossidazione e di riduzione, calcolando il numero di elettroni ceduti da chi si ossida ed acquistati da chi si riduce;
- calcolare il minimo comune multiplo tra il numero degli elettroni ceduti ed il numero degli elettroni acquistati;
- moltiplicare gli elementi che si ossidano e quelli che si riducono per dei coefficienti tali da far circolare lo stesso numero di elettroni.
- Sommare membro a membro le due semireazioni, associando agli elementi che si ossidano o che si riducono gli altri atomi presenti nei composti di partenza, in modo tale da riavere le sostanze presenti nella reazione originaria.
A questo punto abbiamo bilanciato gli elementi che partecipano alla reazione (ovvero che si ossidano o si riducono), resta ora da bilanciare gli elementi che non partecipano alla reazione (ovvero che non variano il loro numero di ossidazione). Ciò deve essere fatto seguendo le ulteriori regole di seguito elencate:
- Se elementi di questo tipo sono presenti all’interno della reazione, si bilanciano prima i metalli, poi i non metalli, poi l’idrogeno;
- Se nella reazione sono presenti composti ionici si deve fare in modo che anche le cariche elettriche siano bilanciate. Ciò in genere viene fatto aggiungendo ioni H+ a sinistra (se la reazione avviene in ambiente acido) oppure, più raramente, aggiungendo ioni OH- a destra (se la reazione avviene in ambiente basico). L’ambiente della reazione deve comunque essere specificato nel testo. Nel bilanciare l’idrogeno si deve ovviamente tener conto anche degli ioni H+ o OH- eventualmente aggiunti.
- Se il bilanciamento è stato fatto correttamente, l’ossigeno deve risultare bilanciato da solo, altrimenti il bilanciamento è sbagliato.
Esempio di bilanciamento di una reazione redox
Data la seguente reazione redox che avviene in ambiente acido: NO3- + Fe2+ è Fe3+ + NO

- si calcolano i numeri di ossidazione:
-
 N si riduce ed Fe si ossida;
N si riduce ed Fe si ossida;
3) si scrivono le semireazioni:
- ogni atomo di azoto scambia 3 elettroni, mentre ogni atomo di ferro ne scambia uno solo.

 si scrivono gli opportuni coefficienti davanti agli elementi che partecipano alla reazione e si sommano membro a membro le due semireazioni:
si scrivono gli opportuni coefficienti davanti agli elementi che partecipano alla reazione e si sommano membro a membro le due semireazioni:
- si aggiungono gli altri elementi chimici in modo da avere le sostanze della reazione di partenza:
NO3- + 3 Fe2+ è 3 Fe3+ + NO
- a questo punto però a sinistra abbiamo 5 cariche positive, mentre a destra ne abbiamo 9, per riequilibrare la situazione aggiungiamo quindi a sinistra 4 ioni H+, poiché la reazione avviene in ambiente acido:
NO3- + 3 Fe2+ + 4 H+ è 3 Fe3+ + NO
- non ci resta ora che bilanciare l’idrogeno e l’ossigeno aggiungendo 2 molecole di acqua a destra:
NO3- + 3 Fe2+ + 4 H+ è 3 Fe3+ + NO + 2H2O
- la reazione è ora completamente bilanciata in modo corretto, poiché anche l’ossigeno bilanciato.
I semielementi di una pila ed il potenziale redox
Come già sappiamo, una reazione di ossidoriduzione può essere divisa in due semi reazioni. Per esempio, la reazione
Sn2+ + Fe → Sn + Fe2+
può essere divisa nelle due seguenti semi reazioni:
Fe → Fe2+ + 2e─ (ossidazione) e Sn2+ + 2e─ → Sn (riduzione)
 Queste debbono necessariamente avvenire nello stesso tempo, perché un elemento si ossida solo se contemporaneamente un altro si riduce e gli elettroni ceduti dal primo sono acquistati dal secondo. In ogni semi reazione compaiono due sostanze, che differiscono tra loro soltanto per uno o più elettroni: una coppia di sostanze di questo tipo viene detta semi elemento di una pila o semplicemente semi elemento e vengo rappresentate nel modo seguente Fe/Fe2+, Sn/Sn2+. I semi elementi vengono chiamati così perché, se accoppiati, possono formare una pila elettrica, cioè un dispositivo all’interno del quale si sviluppa una reazione chimica che produce energia elettrica. Le due sostanze che formano un semi elemento sono i reagenti ed i prodotti di una reazione di ossidoriduzione ed in questo senso assomigliano ad una coppia coniugata acido – base, che è formata invece da due sostanze che differiscono tra loro solo per uno ione H+.
Queste debbono necessariamente avvenire nello stesso tempo, perché un elemento si ossida solo se contemporaneamente un altro si riduce e gli elettroni ceduti dal primo sono acquistati dal secondo. In ogni semi reazione compaiono due sostanze, che differiscono tra loro soltanto per uno o più elettroni: una coppia di sostanze di questo tipo viene detta semi elemento di una pila o semplicemente semi elemento e vengo rappresentate nel modo seguente Fe/Fe2+, Sn/Sn2+. I semi elementi vengono chiamati così perché, se accoppiati, possono formare una pila elettrica, cioè un dispositivo all’interno del quale si sviluppa una reazione chimica che produce energia elettrica. Le due sostanze che formano un semi elemento sono i reagenti ed i prodotti di una reazione di ossidoriduzione ed in questo senso assomigliano ad una coppia coniugata acido – base, che è formata invece da due sostanze che differiscono tra loro solo per uno ione H+.
 Le analogie con le reazioni acido – base non si fermano qui. Ricorderete infatti che una reazione acido – base è tanto più spostata a destra, quanto maggiore è la forza sia dell’acido (nel cedere il protone), che della base (nell’accettarlo). A causa di ciò, per trovare una misura assoluto della forza degli acidi e delle basi è stato necessario confrontare tutte le specie chimiche con una sostanza di riferimento: l’acqua. Anche nelle reazioni redox l’equilibrio è spostato tanto più a destra quanto maggiore è la tendenza degli ossidanti a ridursi e dei riducenti ad ossidarsi. Ancora una volta è quindi difficile misurare una tendenza assoluta di una sostanza ad ossidare o a ridurre. Poiché però un forte ossidante sarà un debolissimo riducente e viceversa, invece di continuare a parlare di tendenza ad ossidare e tendenza a ridurre, parleremo solo di tendenza ad ossidare; i riducenti più forti saranno ovviamente gli ossidanti più deboli. Inoltre, anche in questo caso, per poter stabilire una tendenza ossidante assoluta è stato preso un semi elemento di riferimento, in questo caso quello formato dalla coppia H/H+, e per effettuare una misura quantitativa della tendenza in esame è stata introdotta una nuova grandezza, denominata potenziale redox E0. Fisicamente E0 è una differenza di potenziale elettrico e si misura in volt.
Le analogie con le reazioni acido – base non si fermano qui. Ricorderete infatti che una reazione acido – base è tanto più spostata a destra, quanto maggiore è la forza sia dell’acido (nel cedere il protone), che della base (nell’accettarlo). A causa di ciò, per trovare una misura assoluto della forza degli acidi e delle basi è stato necessario confrontare tutte le specie chimiche con una sostanza di riferimento: l’acqua. Anche nelle reazioni redox l’equilibrio è spostato tanto più a destra quanto maggiore è la tendenza degli ossidanti a ridursi e dei riducenti ad ossidarsi. Ancora una volta è quindi difficile misurare una tendenza assoluta di una sostanza ad ossidare o a ridurre. Poiché però un forte ossidante sarà un debolissimo riducente e viceversa, invece di continuare a parlare di tendenza ad ossidare e tendenza a ridurre, parleremo solo di tendenza ad ossidare; i riducenti più forti saranno ovviamente gli ossidanti più deboli. Inoltre, anche in questo caso, per poter stabilire una tendenza ossidante assoluta è stato preso un semi elemento di riferimento, in questo caso quello formato dalla coppia H/H+, e per effettuare una misura quantitativa della tendenza in esame è stata introdotta una nuova grandezza, denominata potenziale redox E0. Fisicamente E0 è una differenza di potenziale elettrico e si misura in volt.
Tutti i semi elementi capaci di ossidare il semi elemento di riferimento avranno potenziale redox positivo e di valore tanto maggiore quanto più grande è la loro forza come ossidanti. Le sostanze invece che vengono ossidate dal semi elemento di riferimento avranno potenziale redox negativo e di valore algebricamente tanto minore quanto più grande è la loro forza come riducenti. I più forte ossidanti avranno quindi i valori maggiori del potenziale redox (E0>0), mentre i più forti riducenti avranno i valori di potenziale redox più bassi (E0<0).
La tabella a lato riporta i valori di E0 di alcuni tra i semi elementi più importanti. Osservandone i valori si scopre che gli elementi degli ultimi gruppi a destra della tabella formano semi elementi con E0 positivo e grande (forti ossidanti), mentre gli elementi dei primi gruppi a sinistra formano semi elementi con E0 negativo e piccolo (forti riducenti).
La conoscenza dei potenziali redox ci fornisce molte utili indicazioni. Continuando l’analogia con le reazioni acido base, ove abbiamo visto che qualsiasi composto si comporta da acido o da base a seconda delle sostanze con cui viene messo a confronto, anche nelle reazioni redox un fortissimo ossidante riuscirà ad ossidare non solo un riducente, ma anche un ossidante più debole. Utilizzando i potenziali redox possiamo dunque capire in che senso è spostata una reazione e quanto realmente essa avvenga o meno.
Possiamo infatti dire che ogni semi elemento tende ad ossidare i semi elementi con E0 minore, mentre tende a ridurre i semi elementi con E0 maggiore. Inoltre la reazione sarà tanto più spostata a destra quanto maggiore è la forza dell’ossidante (E0 più grande) e maggiore è la forza del riducente (E0 più piccolo). La tendenza ad avvenire di una reazione redox può essere misurata dal valore della seguente differenza: DE0 = E0ossidante - E0 riducente, ove ogni valore del potenziale redox deve essere preso col proprio segno.
Consideriamo di nuovo la reazione
Sn2+ + Fe → Sn + Fe2+
per capire in che senso è spostato l’equilibrio dobbiamo esaminare le due semi reazioni già viste
Fe → Fe2+ + 2e─ e Sn2+ + 2e─ → Sn
Nella prima troviamo le sostanze che formano il semi elemento Fe/Fe2+, il cui potenziale redox è –0.44 volt, nella seconda troviamo invece il semi elemento Sn/Sn2+, il cui potenziale redox è –0.16 volt. Dall’analisi di questi valori ricaviamo subito che lo stagno si riduce, perché ha potenziale maggior, mentre il ferro si ossida, perché ha potenziale minore: l’equilibrio della reazione è dunque spostato a destra. La differenza di potenziale associato a questa reazione è ÄE0 = – 0.16 –(– 0.44) = + 0.2
8 volt.
Consideriamo invece la reazione Sn + 2HCl → SnCl2 + H2
che è scomponibile nelle due semi reazioni
Sn → Sn2+ + 2e─ e H+ + 1 e─ → H
Alla prima, come già visto, corrisponde il semi elemento Sn/Sn2+, il cui potenziale redox è –0.16 volt; alla secondo corrisponde invece il semi elemento H/H+, il cui potenziale redox è 0.00 volt Stavolta lo stagno si ossida, perché ha potenziale minore dell’idrogeno, mentre la differenza di potenziale associata a questa reazione è DE0 = 0. –(– 0.16) = + 0.16 volt.
Consideriamo poi la reazione Fe + 2HCl → FeCl2 + H2
scomponibile nelle due semi reazioni
Fe → Fe2+ + 2e- e H+ + 1 e- → H
Il semi elemento della prima reazione, Fe/Fe2+, ha potenziale redox di –0.44 volt, mentre io semi elemento della seconda, H/H+, ha potenziale redox di 0.00 volt. Nuovamente l’idrogeno si riduce ed il ferro si ossida, mentre la differenza di potenziale è DE0 = 0. –(– 0.44) = + 0.44 volt. La reazione è più spostata a destra della precedente perché, a parità di ossidante, il ferro è un riducente più forte dello stagno.
Il semi elemento H/H+ è contenuto in tutti gli acidi, ed avendo potenziale redox di 0.00 volt, ossida tutti i metalli con E0> 0 (ferro, stagno, piombo, zinco ecc.), mentre non può ossidare il rame (DE0 =+0.34 volt) e l’argento (DE0 =+0.81 volt); per questo motivo il rame e l’argento sono considerati metalli semi nobili.
L’ossigeno (O/O2-) ha un potenziale altissimo (DE0 =+1.29 volt) ed ossida dunque anche i metalli semi nobili, mentre non può ossidare l’oro (Au/Au+++ DE0 =+1.50), che per questo è definito metallo nobile.
Alcuni esempi pratici di reazione redox
Nella vita di tutti i giorni il temine ossidare viene talvolta utilizzato come sinonimo di arrugginire. L’uso è corretto, perché l’arrugginimento del ferro è proprio dovuto alla sua ossidazione da parte dell’ossigeno atmosferico. Anche gli altri metalli non nobili si ossidano, ricoprendosi di una patina formata dal loro ossido. In alcuni casi però questa patina è impermeabile e quindi protegge il metallo non ossidato sottostante dall’azione dell’ossigeno atmosferico: tale fenomeno, detto passivazione, viene largamente utilizzato per proteggere dall’ossidazione oggetti e superfici metalliche.
Un esempio di applicazione della passivazione è la latta, largamente utilizzata nella produzione di barattoli e scatole di metallo, anche per uso alimentare; il cui nome corretto è in verità banda stagnata. Essa infatti è costituita da una lamina di ferro (la banda), ricoperta da una pellicola di stagno; poiché lo stagno si passiva, protegge il ferro sottostante dalla ossidazione. Attenzione però, se con un graffio si toglie la stagnatura, esponendo all’aria il ferro sottostante, questo si ossida molto velocemente, perché si innesca una reazione redox con lo stagno che ha potenziale maggiore del ferro, come abbiamo visto nell’esempio della pagina precedente.
Un’altra applicazione pratica degli stessi principi è la zincatura del ferro, che consiste nel ricoprire l’oggetto da proteggere di uno strato, anche non continuo, di zinco. Questo metallo ha un potenziale minore del ferro (Zn/Zn2+ DE0 =– 0.76) ed innesca con esso una reazione redox del tipo
Fe2+ + Zn → Fe + Zn2+ che impedisce al ferro di ossidarsi. Col tempo però lo zinco della zincatura si consuma e la protezione cessa.
Sullo stesso principio si basano anche le protezioni catodiche, che si impiegano per proteggere dall’ossidazione grandi strutture in ferro come tralicci o simili. La struttura da proteggere viene collegata, tramite un conduttore di corrente, con un pezzo di metallo dotato di potenziale redox molto minore del ferro, come ad esempio il magnesio (Mg/Mg2+ DE0 =– 2.37). Il contatto tra i due metalli innesca la reazione Fe2+ + Mg → Fe + Mg2+ che impedisce l’ossidazione del ferro. Col tempo il pezzo di magnesio si consuma e deve essere sostituito.
Le pile elettriche
Consideriamo adesso la reazione Cu2+ + Zn → Cu + Zn2+
scomponibile nelle due semi reazioni
Zn → Zn2+ + 2e- e Cu2+ + 2e- → Cu
nelle quali lo zinco si ossida, perché ha potenziale minore, mentre il rame si riduce, perché ha potenziale maggiore. Per far avvenire la reazioni complessiva possiamo preparare una soluzione di solfato rameico (CuSO4), che in acqua si decompone nel modo seguente: CuSO4→ Cu2+ + SO42-. Se inseriamo nella soluzione una sbarretta di zinco metallico abbiamo entrambi i reagenti e la reazione inizia a svilupparsi. Lo zinco della lamina va in soluzione, sotto forma di ioni Zn2+, mentre lascia sulla lamina due elettroni, utilizzati dagli ioni Cu2+, che si depongono sulla lamina di zinco in forma di rame metallico. La reazione è spontanea, perché i prodotti sono più stabili dei reagenti, e quindi esoergonica, mentre l'energia prodotta si disperde nel recipiente sotto forma di calore.
La medesima reazione può avvenire però anche a distanza, se si fanno avvenire le due semi reazioni in recipienti separati, ma collegato da un conduttore elettrico. In un recipiente si prepara dunque una soluzione di solfato di zinco, che si decompone così: ZnSO4→ Zn2+ + SO42-, e successivamente ci si inserisce una sbarretta di zinco metallico. In un altro recipiente si mette invece una soluzione di solfato rameico, che si decompone nel modo seguente: CuSO4→ Cu2+ + SO42-, all’interno della quale si inserisce poi una lamina di rame metallico . In ognuno dei due recipienti abbiamo così un semi elemento; se le due lamine metalliche vengono collegate con un filo conduttore , nel primo recipiente avviene la reazione di ossidazione Zn → Zn2+ + 2e-, mentre nel secondo avviene la reazione di riduzione Cu2+ + 2e- → Cu. Nel primo recipiente lo zinco metallico va in soluzione come ioni Zn2+, gli elettroni che si liberano corrono lungo la lamina ed il conduttore esterno fino a raggiungere l’altra lamina, dove servono per far avvenire la reazione di riduzione ed il rame metallico si deposita sulla sua lamina. Lungo il conduttore esterno transitano degli elettroni, cioè una corrente elettrica, che può essere rilevata dall’accendersi di una lampadina posta lungo il conduttore medesimo. Ciascuno dei due recipienti costituisce un semi elemento , così chiamato proprio perché rappresenta la metà di una pila. Il semi elemento con lo zinco, all’interno del quale avviene la reazione di ossidazione, si chiama anodo, mentre il semi elemento col rame, ove avviene la reazione di riduzione, si chiama catodo. Mano a mano che la reazione procede all’anodo lo zinco metallico va in soluzione ed aumenta la concentrazione di ioni Zn2+, mentre al catodo il rame si depone sulla sbarretta e la concentrazione degli ioni Cu2+ diminuisce. Dopo un certo tempo, poiché all’anodo si ha un eccesso di cariche positive, mentre al catodo le cariche positive sono in difetto, si crea uno squilibrio elettrico tra i due semi elementi che richiama indietro gli elettroni, arrestando così la reazione. Per ovviare a ciò si utilizza un ponte salino, cioè un tubicino ripiegato ad u e riempito di una sostanza ionica che scambia ioni tra i due semi elementi, ristabilendo così l’equilibrio ionico ed impedendo che la reazione si arresti. Con questo accorgimento la reazione procede fino all’esaurimento dei reagenti (gli ioni Cu2+ o la sbarretta di zinco) e la pila si scarica. L’anodo è il polo negativo della pila, perché è quello dove si originano le cariche negative (gli elettroni), mentre il catodo è il polo positivo, perché è quello che richiama le cariche negative.
Se al posto della lampadina inseriamo un voltmetro, cioè uno strumento capace di misurare una differenza di potenziale elettrico , misuriamo la differenza di potenziale tra i due poli della pila, chiamata anche Forza Elettromotrice (F.E.M.) della pila stessa. La FEM può essere presa come una misura della tendenza degli elettroni a spostarsi tra i due poli, quindi come la loro tendenza a passare dal riducente all’ossidante ovvero come la tendenza della reazione ad avvenire. In verità la FEM è ciò che finora abbiamo chiamato DE0, quindi possiamo dire che FEM = E0ossidante - E0 riducente. Essa misura quindi la tendenza degli elettroni a spostarsi tra le due sostanze, senza riuscire però a distinguere la tendenza del riducente a cede da quella dell’ossidante a prendere. Come sappiamo il problema si risolve prendendo un semi elemento di riferimento (H/H+), cui si assegna E0=0. Il semi elemento di riferimento, detto anche elettrodo standard è costituito da un filamento di platino, che non partecipa alla reazione, immerso in una soluzione acida (contenente cioè ioni H+) e su cui si fa gorgogliare idrogeno gassoso. Per stabilire se una qualsiasi coppia redox è ossidante o riducente si costruisce con essa un semi elemento e lo si collega in una pila all’elettrodo standard; con un amperometro si misura poi in che senso si spostano gli elettroni.- Se gli elettroni si spostano dalla semi cella di misura all’elettrodo standard, la coppia redox della semi cella si ossida ed è quindi un riducente; ad essa si assegna un E0<0.
- Se gli elettroni si spostano dall’elettrodo standard alla semi cella di misura, la coppia redox di quest’ultima si riduce ed è quindi un ossidante; ad essa si assegna un E0>0.
Con un voltmetro si misura poi la FEM della pila, che in questi casi, poiché l’elettrodo standard ha E0=0, fornisce direttamente il potenziale redox della semi cella di misura. In effetti i potenziali elencati nella tabella di pagina 7 sono stati misurati sperimentalmente in questo modo.
La pila Leclanchè
Le pile come quella sopra descritta sono difficili da trasportare, in quanto le soluzioni possono facilmente rovesciarsi e mescolarsi. Molti più pratiche e maneggevoli sono le pile a secco, in cui le soluzioni imbevono materiali adsorbenti. La pila Leclanchè, comunemente utilizzata negli apparecchi portatili, appartiene a questa tipologia. Essa è costituita da un involucro esterno di zinco metallico, che funge da polo negativo, rivestito internamente da un cartoncino imbevuto di cloruro di zinco (ZnCl2): in questi modo abbiamo il semi elemento Zn/Zn2+: Procedendo verso l’interno troviamo uno strato di materiale inerte imbevuto di cloruro di ammonio (NH4Cl), poi un strato di pasta di biossido di manganese (MnO2) ed infine una sbarretta di grafite che funge da polo positivo. Collegando i due poli attraverso un conduttore esterno gli atomi di zinco dell’involucro si ossidano secondo la reazione Zn → Zn2+ + 2e-; i due elettroni così prodotti raggiungono la sbarretta di grafite attraverso il conduttore esterno e migrano poi verso la parte interna della pila, ove riducono il biossido di manganese secondo la reazione: 2MnO2+2NH4++2H2O+2e-→2Mn(OH)3+2NH3
 La corrente che attraversa il conduttore viene naturalmente utilizzata per azionare un dispositivo elettrico (radio, mangianastri ecc.); mano a mano che la pila funziona l’involucro esterno si consuma e si assottiglia. Quando la pila si è scaricata l’involucro diventa fragile e possono fuoriuscire dei liquidi altamente tossici e corrosivi: non conviene tenere quindi le pile scariche negli apparecchi ed è necessario raccoglierle in modo differenziato. Le pile di questo tipo hanno una differenza di potenziale di 1.5 volt, se si vogliono differenze di potenziale maggiori, basta accoppiare tra loro più pile.
La corrente che attraversa il conduttore viene naturalmente utilizzata per azionare un dispositivo elettrico (radio, mangianastri ecc.); mano a mano che la pila funziona l’involucro esterno si consuma e si assottiglia. Quando la pila si è scaricata l’involucro diventa fragile e possono fuoriuscire dei liquidi altamente tossici e corrosivi: non conviene tenere quindi le pile scariche negli apparecchi ed è necessario raccoglierle in modo differenziato. Le pile di questo tipo hanno una differenza di potenziale di 1.5 volt, se si vogliono differenze di potenziale maggiori, basta accoppiare tra loro più pile. La pila a bottone
La pila a bottoneUn altro tipo di pila a secco è quella denominata Mallory o a bottone, utilizzata negli orologi, nelle macchine fotografiche ecc. Una pila a bottone è formata da un coperchio metallico, che funge da polo negativo, da un disco di polvere di zinco, da un disco imbevuto di idrossido di potassio, da un terzo disco di ossido di mercurio ed, infine, da un contenitore metallico che funge da polo positivo. Collegando i due poli con un circuito esterno lo zinco si ossida secondo la reazione
 Zn → Zn2+ + 2e-, mentre il mercurio si riduce secondo la reazione Hg2+ + 2e-→ Hg.
Zn → Zn2+ + 2e-, mentre il mercurio si riduce secondo la reazione Hg2+ + 2e-→ Hg. Le pile a combustibile
Nelle pile di questo tipo l’energia chimica di combustibili viene convertita direttamente in energia elettrica, senza alcuna combustione. Esse sono costituite da due elettrodi A negativo e B positivo, separati da una zona contenente un elettrolita (ad es. KOH), attraverso la quale migrano degli ioni ma non gli elettroni. Nel caso della pila ad idrogeno ed ossigeno, i due gas vengono forniti direttamente agli elettrodi, ove avvengono le due reazioni:
in A 2H2 + 4OH- → 4 H2O + 4e- (ossidazione)
in B 4e-+ O2 + 2 H2O → 4OH- (riduzione)
sommando membro a membro le due reazioni si ottiene la reazione complessiva 2H2+ O2→2 H2O, che abbiamo già visto essere quella relativa alla combustione dell’idrogeno. Gli elettroni prodotti in A transitano nel circuito esterno fino a B, ove riducono l’ossigeno, mentre i 4 OH- prodotti in B chiudono il circuito migrando verso A attraverso l’elettrolita. Queste pile hanno un rendimento molto elevato, sono utilizzate per questo sui veicoli spaziali, e producendo come gas di scarico solo vapore acqueo sono veramente ecologiche.Gli accumulatori
Sono dispositivi capaci di trasformare energia chimica in energia elettrica e viceversa, sono cioè ricaricabili. In un recipiente contenente una soluzione acquosa di H2SO4 sono immerse due piastre di piombo, traforate in modo da formare delle cellette. La piastra che funge da polo negativo (anodo) ha le cellette ripiene di piombo metallico, che si ossida producendo ioni Pb2+ e due elettroni; gli ioni così prodotti restano nelle cellette perché si legano agli ioni SO42- presenti in soluzione, formando solfato piomboso PbSO4. Attraverso il conduttore esterno i due elettroni passano da questa piastra all’altra, che funge da polo positivo (catodo), le cui cellette sono piene di ossido piombico (PbO2). Quest’ultimo viene ridotto a ioni Pb2+, i quali, a loro volta si legano agli ioni SO42- della soluzione, formando altro solfato piomboso che, essendo insolubile, resta nelle cellette. Le due reazioni che si sviluppano sono le seguenti:
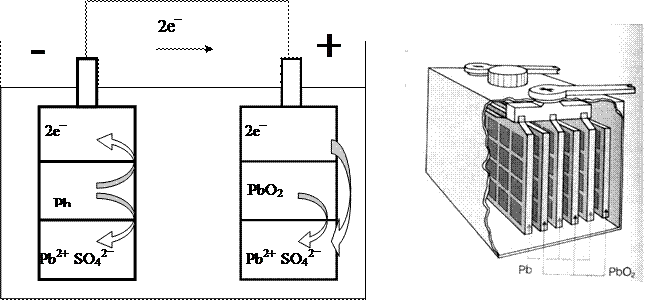 all’anodo Pb + SO42-→ PbSO4 + 2e-
all’anodo Pb + SO42-→ PbSO4 + 2e-
al catodo PbO2 + SO42- + 4H+ + 2e-→ PbSO4 + 2H2O
Mentre la reazione complessiva è la seguente: Pb + PbO2 + 2 H2SO4 → PbSO4 + 2H2O (1)
I due semi elementi in gioco in questa pila sono Pb/ Pb2+ e Pb2+/ Pb4+ e la FEM è di 2 volt. Collegando in serie tra loro più piastre, in genere 6 o 12, si ottiene una batteria da auto con una FEM complessiva, rispettivamente, di 12 o 24 volt. Via via che la batteria produce corrente elettrica l’acido solforico si consuma e si produce acqua, con una conseguente diminuzione della densità della soluzione ed in effetti un metodo per misurare la carica di una batteria è proprio quello di rilevare la densità della soluzione interna.
Se la batteria viene collegata ad un generatore di corrente, come la dinamo della macchina, la reazione (1) avviene in senso inverso e la batteria si ricarica, trasformando energia elettrica in energia chimica. Nelle auto le batterie vengono utilizzate come pile nell’avviamento del motore, mentre vengono ricaricate quando il motore è acceso.I conduttori elettrici
Studiando il legame metallico abbiamo visto che i metalli conducono la corrente elettrica perché i loro elettroni più esterni sono libri di muoversi attraverso il metallo. Con il loro spostamento tali elettroni assicurano il passaggio della carica elettrica proprio della corrente, mentre nel metallo non si verifica alcun tipo di reazione chimica. Sappiamo inoltre che l’acqua pura non conduce la corrente; ma se tuttavia vi sciogliamo un composto ionico, come un sale, o un composto covalente polare, come un acido, la corrente circola nella soluzione. Tali sostanze si dicono elettroliti e le loro soluzioni si definiscono soluzioni elettrolitiche; al loro interno il passaggio della corrente non è dovuto al movimento di elettroni, come nei metalli, bensì allo spostamento degli ioni. La quantità di corrente che transita in tali soluzioni è funzione della quantità di ioni presenti al loro interno; essa dipende quindi dalla concentrazione dell’elettrolita e, se questo è un acido, anche dalla sua forza. Entro le soluzioni elettrolitiche, infine, il passaggio della corrente determina lo svilupparsi di reazioni chimiche.
L’elettrolisi
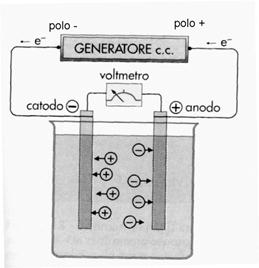 Prepariamo una soluzione di HCl ed immergiamoci dentro due sbarrette di una metallo inerte, collegandole poi ai due poli di una pila o di un generatore di corrente. Le due sbarrette si chiamano elettrodi, perché conducono la corrente; quella collegata al polo negativo si caricherà negativamente, mentre quella collegata al polo positivo si caricherà positivamente. Nella soluzione di HCl sono presenti ioni H+ (cationi) e ioni Cl- (anioni); sotto l’effetto delle cariche elettriche distribuite sui due elettrodi i cationi, carichi positivamente, saranno attratti dall’elettrodo negativo, che per questo si dice catodo, viceversa gli anioni, carichi negativamente, saranno attratti dall’elettrodo positiva, che pertanto viene chiamato anodo.
Prepariamo una soluzione di HCl ed immergiamoci dentro due sbarrette di una metallo inerte, collegandole poi ai due poli di una pila o di un generatore di corrente. Le due sbarrette si chiamano elettrodi, perché conducono la corrente; quella collegata al polo negativo si caricherà negativamente, mentre quella collegata al polo positivo si caricherà positivamente. Nella soluzione di HCl sono presenti ioni H+ (cationi) e ioni Cl- (anioni); sotto l’effetto delle cariche elettriche distribuite sui due elettrodi i cationi, carichi positivamente, saranno attratti dall’elettrodo negativo, che per questo si dice catodo, viceversa gli anioni, carichi negativamente, saranno attratti dall’elettrodo positiva, che pertanto viene chiamato anodo.
Al catodo gli elettroni sulla superficie dell’elettrodo, provenienti dal polo negativo del generatore, vengono ceduti agli ioni H+ secondo la reazione: 2 H+ + 2e-→ H2; l’idrogeno si riduce e si sviluppa in forma gassosa.
 All’anodo invece gli ioni Cl- cedono ciascuno un elettrone all’elettrodo positivo, secondo la reazione
All’anodo invece gli ioni Cl- cedono ciascuno un elettrone all’elettrodo positivo, secondo la reazione
2Cl-→ Cl2 + 2e-; il cloro si ossida e si sviluppa in forma gassosa. Il passaggio degli elettroni dal catodo agli ioni H+ e dagli ioni Cl- all’anodo, chiude il circuito elettrico, spiegando la conduzione di corrente attraverso la soluzione. Complessivamente infatti un elettrone si è spostato dal catodo all’anodo. Sommando membro a membro le reazioni che avvengono agli elettrodi otteniamo la reazione complessiva 2 H++2Cl-→ H2 + Cl2, nella quale l’idrogeno si riduce ed il cloro si ossida. Essa non è spontanea, perché è spontanea la reazione opposta: la coppia Cl/Cl- ha infatti potenziale standard E0> della coppia H/H+ . Nel fenomeno descritto, che prende il nome di elettrolisi, l’energia elettrica è servita per far avvenire una reazione chimica endoergonica; abbiamo quindi trasformato energia elettrica in energia chimica, l’opposto di ciò che avveniva in una pila.
Sia nell’elettrolisi che nella pila abbiamo un anodo ed un catodo; in entrambi i casi all’anodo avviene l’ossidazione, mentre al catodo avviene la riduzione. Per ricordare meglio questo fatto basterà rammentare che la parola anodo e la parola ossidazione cominciano per vocale, mentre tanto catodo quanto riduzione cominciano per consonante. Nella pila una reazione redox produce energia elettrica; il catodo è il polo positivo, mentre l’anodo è quello negativo. Nell’elettrolisi invece l’energia elettrica fa avvenire una reazione redox; il catodo è l’elettrodo negativo, mentre l’anodo è l’elettrodo positivo.L’elettrolisi dell’acqua
Sottoponendo ad elettrolisi dell’acqua pura abbiamo lo sviluppo delle seguenti reazioni:
all’anodo 2 H2O → O2 + 4 H+ + 4e- (ossidazione) (1)
al catodo 2 H2O + 2e- → H2 + 2OH- (riduzione) (2)
Poiché le due reazioni sono contemporanee e collegate, è necessario bilanciare la (2) nel modo seguente: 4 H2O + 4e- → 2H2 + 4OH- (3).
Sommando membro a membro la (1) e la (3) si ottiene la reazione complessiva di elettrolisi dell’acqua 6 H2O → O2 + 2H2 + 4OH- + 4H+
 Poiché gli H+ reagiscono con gli OH- per ridare acqua, secondo la reazione: H+ + OH- → H2O, alla fine la reazione complessiva diventa:
Poiché gli H+ reagiscono con gli OH- per ridare acqua, secondo la reazione: H+ + OH- → H2O, alla fine la reazione complessiva diventa:
2H2O → O2 + 2H2
esattamente l’opposto della reazione di formazione dell’acqua dalla combustione dell’idrogeno.
L’elettrolisi dell’acqua viene fatta avvenire in un apparecchio, chiamato voltametro, rappresentato nella figura a fianco. Per consentire all’acqua di condurre la corrente vi viene aggiunta una piccola quantità di acido, senza la quale la reazione non procede.L’elettrolisi di una soluzione di cloruro di sodio
Il cloruro di sodio in soluzione acquosa da luogo alla reazione: NaCl →Na+ + Cl-. Sottoponendo ad elettrolisi una tale soluzione si verifica la consueta migrazione degli ioni verso i due elettrodi: Cl- (anioni) verso l’anodo ed Na+ (cationi) verso il catodo.
All’anodo si verifica la reazione 2Cl-→ Cl2 + 2e- con la quale il cloro si ossida.
 . Quindi, mentre all’anodo si sviluppa cloro gassoso, al catodo si sviluppa idrogeno gassoso ed in soluzione rimangono ioni Na+ e OH-, che, facendo evaporare l’acqua, danno luogo ad NaOH. L’elettrolisi del cloruro di sodio assume una notevole importanza industriale, in quanto NaCl si ricava facilmente per evaporazione dell’acqua di mare, mentre i tre prodotti che si ottengono (H2 gassoso, Cl2 gassoso e NaOH, noto commercialmente come soda caustica), sono largamente impiegati nell’industria chimica.
. Quindi, mentre all’anodo si sviluppa cloro gassoso, al catodo si sviluppa idrogeno gassoso ed in soluzione rimangono ioni Na+ e OH-, che, facendo evaporare l’acqua, danno luogo ad NaOH. L’elettrolisi del cloruro di sodio assume una notevole importanza industriale, in quanto NaCl si ricava facilmente per evaporazione dell’acqua di mare, mentre i tre prodotti che si ottengono (H2 gassoso, Cl2 gassoso e NaOH, noto commercialmente come soda caustica), sono largamente impiegati nell’industria chimica. Elettrolisi di elettroliti fusi
Dunque dall’elettrolisi di una soluzione di NaCl non è possibile ricavare Na metallico, perché al catodo, invece di questo metallo, si riduce l’acqua, a causa del suo potenziale redox maggiore. Per ottenere la riduzione del sodio, e quindi l’ottenimento di questo metallo allo stato elementare, è necessario sottoporre ad elettrolisi NaCl allo stato fuso. Il sale, mischiato ad altri componenti che ne abbassano la temperatura di fusione, viene fuso a circa 600°C e quindi sottoposto ad elettrolisi, con lo sviluppo delle seguenti reazioni:
all’anodo (ossidazione) 2Cl-→ Cl2 + 2e- al catodo (riduzione) Na+ + e- → Na
il cloro gassoso, come già visto, si sviluppa in bolle e viene raccolto, mentre il sodio metallico galleggia sull’elettrolita fuso e viene così raccolto.
Questo processo elettrochimico, noto come riduzione catodica, è impiegato industrialmente per ricavare allo stato elementare diversi metalli con un potenziale redox particolarmente basso, tra cui, oltre al sodio, troviamo il litio, il potassio, il magnesio, il calcio e l’alluminio . Il metodo venne messo a punto agli inizi dell’800 da un chimico inglese, Humphrey Davy, che ricavò per la prima volta i metalli elencati allo stato elementare, ad eccezione però dell’alluminio, i cui composti richiedevano temperature troppo elevate per la fusione. Nel XIX° secolo quindi non era ancora possibile ricavare in modo semplice alluminio elementare, questo metallo era dunque raro e, pertanto molto costoso, tant’è che i ricchi dell’epoca ostentavano la loro ricchezza pranzando con posate di alluminio. A ristabilire un po’ di giustizia sociale pensò un giovane chimico inglese, Martin Hall, che, alla fine dell’800 trovo il modo di abbassare la temperatura di fusione dell’ossido di alluminio, aprendo la strada alla produzione elettrolitica di questo metallo oggi largamente diffuso.La galvanostegia
Come ultima applicazione dei fenomeni elettrochimici citiamo questo procedimento utilizzato per ricoprire oggetti metallico con uno strato molto sottile di un altro metallo, allo scopo di proteggere l’oggetto medesimo dalla corrosione o di renderlo più prezioso. Immaginiamo, ad esempio, di voler argentare una moneta, ricoprendola con un sottilissimo strato di argento. Si prepara una soluzione di nitrato di argento (AgNO3), all’interno della quale si introduce una sbarretta di argento e la moneta; colleghiamo poi entrambi con un generatore di corrente continua: la sbarretta col polo positivo e la moneta col polo negativo. Quando facciamo passare corrente, la sbarretta di argento diventa l’anodo della cella elettrolitica, ove il metallo passa in soluzione sotto forma di ioni Ag+, mentre la moneta diventa il catodo ove gli ioni Ag+ si riducono ad argento metallico che si deposita sulla moneta stessa. Le reazioni che si verificano agli elettrodi sono dunque le seguenti:
all’anodo (ossidazione) Ag → Ag+ + e- al catodo (riduzione) Ag+ + e- → Ag
Con l’aiuto della corrente abbiamo quindi fatto passare degli atomi di argento dalla sbarretta alla moneta. Procedimenti analoghi possono essere utilizzati anche per zincare, stagnare, dorare, cromare oggetti metallici di vario tipo.
La differenza di potenziale elettrico tra due punti può essere immaginata come l’energia posseduta da un elettrone che si sposta tra di essi oppure come la tendenza dell’elettrone a spostarsi tra i due punti. Essa però può essere anche paragonata alla differenza di livello tra due recipienti di acqua: se la differenza è grande, molta acqua passa da un recipiente all’altro prima che si raggiunga lo stesso livello in entrambi. Se viceversa la differenza è piccola, lo stesso livello viene raggiunto col passaggio di poca acqua.
Fonte: http://www.liceodavincifi.it/_Rainbow/Documents/ELETTROCHIMICA2.doc
Elettrochimica
L'elettrochimica studia i processi di trasformazione di energia chimica di legame in energia elettrica e viceversa.
Abbiamo già osservato come una reazione redox sia una reazione in cui una sostanza si ossida, cedendo elettroni ad un'altra sostanza che, acquistandoli, si riduce.Una reazione redox è spontanea quando gli elettroni passano da una sostanza dove si trovano ad un livello energetico più elevato e quindi meno stabile, ad una sostanza dove, essendo trattenuti più fortemente, si trovano in una situazione di maggior stabilità.
Esiste quindi una differenza di potenziale tra la specie che si ossida e quella che si riduce e gli elettroni possono muoversi spontaneamente in risposta a tale gradiente.
Ora, potendo far avvenire una reazione spontanea di ossido-riduzione non direttamente, ma costringendo gli elettroni a passare attraverso un filo metallico, si potrà sfruttare tale differenza di potenziale per produrre una corrente elettrica. In altre parole potremo trasformare energia potenziale (energia chimica) nell'energia cinetica degli elettroni in moto (energia elettrica).
Tale processo può essere realizzato tramite un dispositivo noto come cella galvanica o pila.Al contrario se vogliamo far avvenire una reazione redox in senso inverso a quello spontaneo, costringendo gli elettroni a muoversi contro un gradiente di potenziale, sarà necessario fornire energia al sistema, trasformando energia elettrica in energia chimica. Tale processo si ottiene nelle reazioni di elettrolisi.
Celle galvaniche o pile
Dunque le celle galvaniche sono dei dispositivi in grado di sfruttare reazioni redox spontanee per trasformare energia chimica in energia elettrica.
Una pila è costituita da due semicelle in cui vengono fatte avvenire separatamente le reazioni di ossidazione e di riduzione.Consideriamo ad esempio il seguente processo spontaneo di ossidoriduzione
Zn + Cu2+ → Zn2+ + Cu
dove lo zinco metallico si ossida in ione zinco perdendo due elettroni secondo la seguente semireazione
Zn → Zn2+ + 2e-
e lo ione rame si riduce a rame metallico acquistando due elettroni secondo la seguente semireazione
Cu2+ + 2e- → Cu
Se vogliamo costruire una pila che si basi su tale reazione è necessario separare le due semireazioni. In ciascuna semicella dovrà dunque essere posto uno dei due metalli in equilibrio con il proprio catione.
La semicella che contiene lo zinco verrà indicata come Zn/Zn2+
La semicella che contiene il rame come Cu2+/Cu.Le specie chimiche che partecipano alla reazione in ciascuna semicella sono dette coppie redox. Ciascuna coppia redox viene sempre convenzionalmente scritta nel senso della reazione di riduzione.
Una pila che utilizza proprio la reazione di ossidoriduzione tra rame e zinco è la pila Daniell.Le coppie redox utilizzate nella pila Daniell sono Cu2+/Cu e Zn2+/Zn.
Nella pila Daniell una barretta di zinco metallico viene immersa in una soluzione di solfato di zinco che, dissociandosi completamente, fornisce gli ioni Zn2+, mentre una barretta di rame metallico viene immersa in una soluzione di solfato rameico che fornisce gli ioni Cu2+.
In questo caso particolare le due barrette metalliche fungono anche da elettrodi (o collettori di elettricità), cioè da supporto per il passaggio degli elettroni da una semicella all'altra quando avviene la reazione. In altre pile, dove le specie chimiche che reagiscono non sono conduttrici, è necessario utilizzare elettrodi inerti (che non partecipano alla reazione) per consentire agli elettroni di muoversi.
Spesso il termine elettrodo viene utilizzato per indicare l'intera semicella.

Finche le due semicelle rimangono separate non si osserva naturalmente alcuna reazione. Ma se colleghiamo con un filo metallico i due elettrodi, lo Zinco, che ha una maggiore tendenza ad ossidarsi rispetto al Rame, perde elettroni. Gli elettroni, passando attraverso il conduttore metallico, vengono attirati dagli ioni rameici che li acquistano e si riducono.
In tal modo dello zinco metallico si ossida e passa in soluzione sotto forma di ione Zn2+ (lo ione zinco è solubile). La soluzione di solfato di zinco si arricchisce di ioni Zn2+ mentre l'elettrodo di Zinco si assottiglia.
Nell'altra semicella invece gli ioni rameici a contatto con l'elettrodo si riducono a rame metallico aderendo all'elettrodo stesso. La soluzione si impoverisce di ioni Cu2+ mentre l'elettrodo aumenta di peso.
Convenzionalmente l'elettrodo al quale avviene la reazione di ossidazione è detto Anodo, mentre quello al quale avviene la reazione di riduzione è detto Catodo.
Poiché in tal caso gli elettroni si muovono dall'anodo al catodo, l'anodo risulta essere l'elettrodo negativo, mentre il catodo è positivo.
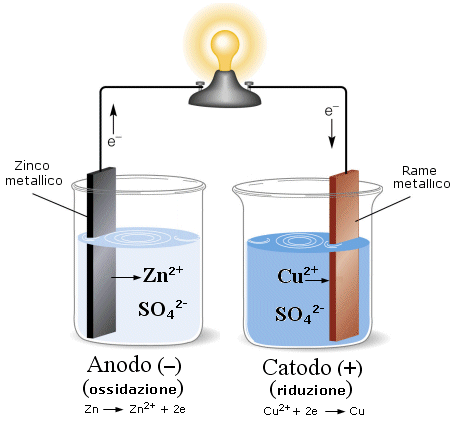
Poiché mentre la reazione procede la soluzione di solfato di zinco tende ad arricchirsi di ioni positivi (Zn2+), mentre la soluzione di solfato rameico tende ad impoverirsi di ioni positivi (Cu2+), le due semicelle perdono la loro elettroneutralità.
La semicella anodica tende a caricarsi positivamente [Zn2+] > [SO42-], mentre la semicella catodica tende a caricarsi negativamente [Cu2+] < [SO42-].In queste condizioni il passaggio di corrente elettrica si esaurirebbe ben presto, in quanto gli elettroni (negativi) dovrebbero allontanarsi da una soluzione positiva, che li attrae, per andare verso una soluzione negativa, che li respinge.
La perdita progressiva dell'elettroneutralità da parte delle due semicelle diventa perciò responsabile di un potenziale elettrico che agisce in senso contrario a quello originario. Quando l'intensità dei due potenziali assume lo stesso valore assoluto, la corrente cessa.Per ovviare a tale inconveniente è quindi necessario garantire l'elettroneutralità delle due soluzioni. Per questo motivo le soluzioni vengono collegate con un dispositivo, detto ponte salino, che fornisce loro ioni di segno opposto.
Un esempio di ponte salino potrebbe essere un tubo contenente una soluzione molto concentrata di un elettrolita forte (ad esempio un sale), i cui ioni non interferiscano chimicamente con le reazioni in corso. Le due estremità del tubo vengono immerse nelle semicelle ed occluse con materiale filtrante in modo che gli ioni di carica opposta possano diffondere lentamente.
Se ad esempio utilizziamo un generico sale BA, in grado di dissociarsi in ioni B+ e A-, gli ioni B+ diffonderanno nella soluzione catodica, mentre gli ioni A- in quella anodica, garantendo l'elettroneutralità.

Il ponte salino può essere sostituito da un setto poroso che divida le due soluzioni (ad esempio porcellana non verniciata) e permetta all'eccesso relativo di ioni SO42- di diffondere verso la soluzione positiva e agli ioni Zn2+ in eccesso di migrare verso la soluzione negativa.

In generale una pila viene schematizzata, scrivendo per prima la reazione anodica come segue
Zn / Zn2+ // Cu2+ / Cu
ANODO PONTE CATODO
dove
Zn / Zn2+ rappresenta la semicella anodica (ossidazione)
// è il simbolo del ponte salino
Cu2+ / Cu rappresenta la semicella catodica (riduzione)
Potenziale di elettrodo
Se inseriamo un voltmetro lungo il filo che unisce i due elettrodi possiamo naturalmente misurare la differenza di potenziale (in Volt) che permette agli elettroni di scorrere lungo il filo da una semicella all'altra.
In modo analogo ai corpi dotati di massa che si muovono (se non vincolati) all'interno di un campo gravitazionale, spostandosi da punti a potenziale gravitazionale maggiore verso punti a gravitazionale minore, così anche le cariche elettriche si muovono sotto l'azione di un campo elettrico che si produce tra punti a diverso potenziale elettrico.
La differenza di potenziale elettrico V tra due punti viene definita come il lavoro necessario per spostare l'unità di carica elettrica da un punto all'altro.
1 Volt (J/C) è la differenza di potenziale esistente tra due punti quando il lavoro necessario per spostare la carica di 1 Coulomb è pari ad un Joule.La differenza di potenziale tra i due elettrodi è quindi una misura della capacità della pila di compiere lavoro ed è quindi anche una misura della forza con cui gli elettroni che si liberano all'anodo durante l'ossidazione vengono spinti, lungo il circuito esterno, verso il catodo.
Sperimentalmente si osserva che tale differenza di potenziale, detta anche forza elettromotrice (f.e.m.) o tensione o voltaggio, dipende esclusivamente dal tipo di reazione redox, cioè dalla natura chimica dei reagenti (maggiore o minore tendenza a perdere o acquistare elettroni), dalla concentrazione delle specie chimiche e dalla temperatura (all'aumentare della temperatura e della concentrazione dei reagenti aumenta la differenza di potenziale).
Durante il funzionamento di una pila i reagenti si trasformano in prodotti di reazione. Poiché quindi le concentrazioni dei reagenti diminuiscono, anche la differenza di potenziale è destinata a calare fino ad azzerarsi. Quando la differenza di potenziale va a zero la pila è scarica ed in tali condizioni non è più in grado di produrre corrente elettrica.
In teoria la forza elettromotrice di una pila si può ottenere come differenza tra il potenziale di un elettrodo ed il potenziale dell'altro elettrodo.
Purtroppo però l'unica grandezza accessibile alla misura è la differenza di potenziale e non il potenziale assoluto di ciascuna semicella.
Infatti in una semicella isolata nella quale la specie ossidata è in equilibrio con la specie ridotta non avviene nessuna reazione. In potenza essa è in grado sia di fornire che di acquistare elettroni, ma in pratica lo fa solo quando viene collegata con un'altra semicella.
Solo collegandolo con un'altra semicella noi possiamo verificare se essa cederà o acquisterà elettroni e potremo effettivamente misurare la differenza di potenziale esistente.E' come se avessimo tre bacini d'acqua disposti lungo un pendio e ci venisse richiesto di prevedere se il bacino centrale (B) è in grado di fornire o di ricevere acqua. E' evidente che per poter rispondere a tale domanda è necessario sapere se esso verrà collegato al bacino A, a monte, (dal quale riceve acqua) o al bacino C, a valle, (al quale cede acqua).

Analogamente, non è possibile misurare la quota di una montagna o di un fondale marino se non decidiamo rispetto a quale riferimento convenzionale (in genere il livello del mare) eseguiamo le misurazioni.
In tal modo non siamo in grado di misurare l'energia potenziale assoluta dell'acqua posta in una certa vasca (Ep = mgh), ma solo la sua energia potenziale rispetto ad un'altra vasca. Ad esempio possiamo misurare il potenziale della vasca A rispetto alle altre vasche conoscendone le differenze di quota (hab e hac) o il potenziale della vasca B rispetto alla vasca C conoscendone la differenza di quota (hbc). In definitiva quindi l'operazione che noi facciamo è una misura della differenza di potenziale tra le vasche.
Proseguendo con il nostro esempio, risulta evidente che se le vasche sono numerose non è comodo misurare i dislivelli tra tutte le coppie di vasche. E' infatti più semplice misurare per tutte le vasche la differenza di potenziale rispetto ad un'unica quota posta convenzionalmente come zero (generalmente il livello del mare, ma volendo qualsiasi altro punto di riferimento è ugualmente accettabile) e assumere tale valore come un valore di potenziale assoluto assegnato convenzionalmente a ciascuna vasca.
In tal modo quando sarà necessario potremmo sempre calcolare la differenza di potenziale tra due vasche utilizzando il loro potenziale calcolato rispetto ad un medesimo punto di riferimento (livello del mare). Si noti che cambiando il punto di riferimento (la quota assunta convenzionalmente come zero), i dislivelli non mutano e le differenze di potenziale rimangono inalterate. La differenza di potenziale tra le due vasche è una misura del lavoro che l'acqua può compiere quando scende dalla vasca più alta a quella più bassa o, il che è lo stesso, del lavoro che è necessario compiere sull'acqua per sollevarla dalla vasca più bassa a quella più alta.In modo del tutto analogo, per non dover effettuare tutte le misurazioni di differenza di potenziale elettrico tra tutte le possibili coppie di semicelle, è stata arbitrariamente scelta una semicella alla quale è stato convenzionalmente assegnato potenziale zero. Poiché la differenza di potenziale varia con la temperatura e la concentrazione dei reagenti, si è inoltre convenzionalmente deciso di effettuare le misure alla temperatura di 25°C e con le specie chimiche alla concentrazione 1M (nel caso di gas pressione parziale di 1 atm).
La semicella assunta come elettrodo di riferimento è costituita dalla coppia redox H+/H2 ed è nota come elettrodo normale ad idrogeno.
Tale semicella viene realizzata immergendo un conduttore inerte (in genere platino) in una soluzione 1 M di ioni H+ ( si utilizza una soluzione 1 N di un acido forte). All'interno della soluzione viene fatto gorgogliare idrogeno gassoso alla pressione di 1 atm, in modo che vada a raccogliersi all'interno di una campana di vetro rovesciata in cui è ospitato l'elettrodo.

Alla semicella così realizzata viene dunque attribuito potenziale zero. Poiché poi si è convenuto di considerare la reazione di riduzione di ciascuna coppia redox, si attribuirà un potenziale positivo a tutte le coppie redox che effettivamente si riducono rispetto all'elettrodo ad idrogeno, ed un potenziale negativo a tutte quelle che si ossidano rispetto all'idrogeno.
Il catodo viene pertanto ad assumere sempre potenziale maggiore dell'anodo.
L'elettrodo ad idrogeno può quindi funzionare, a seconda delle coppie redox con cui viene messo in contatto, sia come anodo che come catodo (naturalmente ciò vale anche per qualsiasi altra coppia redox, che può ridursi o ossidarsi a seconda dei casi).
Deve essere chiaro che il potenziale di una certa semicella non è un potenziale assoluto, ma una differenza di potenziale con l'elettrodo normale ad idrogeno.
I valori in tal modo misurati per ogni coppia redox prendono il nome di Potenziali normali o standard di elettrodo (E°).Se ad esempio poniamo la coppia redox Zn2+/Zn a contatto con l'elettrodo normale ad idrogeno, possiamo misurare una differenza di potenziale di 0.76 V.
Poiché lo zinco si ossida cedendo elettroni agli ioni H+ che si riducono ad idrogeno gassoso (possiamo verificarlo osservando che il pH della soluzione catodica aumenta), lo zinco funge da anodo mentre l'idrogeno da catodo. Dobbiamo pertanto assegnare alla coppia redox Zn2+/Zn un potenziale negativo pari a E° = - 0.76 V (il segno negativo ci informa che rispetto all'idrogeno la reazione di riduzione non è spontanea).
Zn / Zn2+ // H+ / H2ANODO CATODO


Se invece poniamo la coppia redox Cu2+/Cu a contatto con l'elettrodo ad idrogeno, possiamo misurare una differenza di potenziale di 0,34 V.
Poiché in questo caso è l'idrogeno che si ossida cedendo elettroni al rame (possiamo verificarlo osservando che il pH della soluzione anodica diminuisce), l'idrogeno funge da anodo mentre il rame da catodo. Dobbiamo pertanto assegnare alla coppia redox Cu2+/Cu un potenziale positivo pari a E° = + 0.34 V (il segno positivo ci informa che rispetto all'idrogeno la reazione di riduzione è spontanea).H2 /H+ // Cu2+ /Cu
ANODO CATODO


Nello stesso modo è possibile misurare i potenziali standard di molte altre coppie redox. Le coppie redox vengono poi ordinate per potenziali standard decrescenti in una tabella detta serie elettrochimica. Nella serie elettrochimica le coppie redox compaiono, come da convenzione, scritte nel senso della reazione di riduzione e i valori di potenziale associati a ciascuna coppia redox devono essere quindi interpretati come potenziali standard di riduzione.
In tal modo il confronto dei valori tabulati ci indica qual è il senso spontaneo della reazione tra due coppie redox, infatti la coppia redox che presenta un potenziale standard di riduzione più elevato presenta anche la maggior tendenza a ridursi.Prese quindi in considerazione due coppie redox, quella a potenziale più elevato subirà la riduzione e fungerà da catodo, mentre quella a potenziale minore subirà l'ossidazione e fungerà da anodo.
La forza elettromotrice della pila così costruita potrà essere prevista calcolando la differenza tra il potenziale standard di riduzione del catodo e quello dell'anodo
f.e.m. = E°catodo - E°anodoAd esempio la forza elettromotrice della pila Daniell è

Naturalmente tale calcolo è possibile anche quando i potenziali standard di due coppie redox sono entrambi positivi o entrambi negativi.
Ad esempio se costruiamo una pila con la coppia redox Zn2+/Zn (E° = - 0,76 V) e Ba2+/Ba (E° = - 2,90 V), in questo caso lo zinco, che presenta potenziale maggiore si riduce e funge da catodo, mentre il bario, a potenziale minore, si ossida e funge da anodo.
La forza elettromotrice di tale pila sarà
Si tenga presente che i potenziali di riduzione riportati nella serie elettrochimica ci permettono, in generale, di fare delle previsioni sulla spontaneità delle reazioni redox. A parità di concentrazione (ricordiamo infatti che il potenziale varia con la concentrazione) ciascuna coppia redox tende ad ossidare le coppie redox a potenziale inferiore, mentre viene ossidata dalle coppie redox a potenziale maggiore.
Ad esempio tutti i metalli che presentano un potenziale di riduzione inferiore a quello dell'idrogeno (e quindi negativo), vengono ossidati (corrosi) da soluzioni 1N di acidi forti.
Quindi mentre il ferro ( ) rimangono inalterati.
) rimangono inalterati.E' bene sottolineare che i potenziali di riduzione riportati nella serie elettrochimica misurano la tendenza della reazione ad avvenire e sono indipendenti dal numero di elettroni scambiati durante la reazione. Tenendo quindi presente che i potenziali sono sempre riferiti ad un singolo elettrone è possibile combinare opportunamente i potenziali di riduzione per ottenere valori relativi a semireazioni che non compaiono nella serie elettrochimica.
Ad esempio
E°
n
nE°
1a reazione
Fe3+ + e → Fe2+
0,77
1
0,77
2a reazione
Fe2+ + 2e → Fe
- 0,44
2
- 0,88
reazione complessiva
Fe3+ + 3e → Fe
3
- 0,11
La terza reazione risulta la somma delle prime due. Quindi, dopo aver moltiplicato ciascun potenziale per il numero di elettroni, si sommano algebricamente i valori ottenuti ottenendo il potenziale della reazione somma, riferito a 3 elettroni. Si calcola infine il potenziale per singolo elettrone, che nel caso particolare vale

Equazione di Nernst
L'equazione di Nernst permette di calcolare il potenziale di una cella in condizioni di concentrazione diverse da quelle standard.
Per una generica coppia redox An+/A, per la quale la reazione di riduzione sia dunque
An+ + ne- → A
L'equazione di Nernst vale

dove
E = potenziale in condizioni di concentrazione non standard
E° = potenziale standard
R = costante dei gas (8,31 J.mol-1.K-1)
T = temperatura assoluta
n = numero di elettroni scambiati
F = Faraday = 96.485 Coulomb (carica portata da una mole di elettroni = NA·Qe = 6,022.1023 . 1,602.10-19)
ln = logaritmo neperiano o naturale (in base e = 2,7183)
[A] = concentrazione della specie ridotta (eventualmente elevata al proprio coeff. stechiometrico)
[An+] = concentrazione della specie ossidata (eventualmente elevata al proprio coeff. stechiometrico)A temperatura di 25°C il rapporto
 è una costante e trasformando il logaritmo neperiano in un logaritmo in base 10 (coefficiente di conversione 2,3), l'equazione diventa
è una costante e trasformando il logaritmo neperiano in un logaritmo in base 10 (coefficiente di conversione 2,3), l'equazione diventa

E' facile verificare che in condizioni standard, poiché [A] = [An+] = 1N, il logaritmo vale zero e quindi
E = E°
Relazione tra kc e f.e.m. (ΔE°)
Mentre una pila fornisce corrente elettrica al suo interno le reazioni di ossidoriduzione procedono facendo variare le concentrazioni dei reagenti. In tal modo i potenziali delle semicelle variano e si modifica pertanto anche la differenza di potenziale o f.e.m.
Riprendiamo in esame la pila Daniell e calcoliamo per ciascuna semicella il potenziale in condizioni di concentrazione diverse da quelle standard.
ANODO

CATODO

Calcoliamo ora la forza elettromotrice f.e.m. in condizioni di concentrazione non standard come differenza di potenziale E tra catodo ed anodo

riordinando
La reazione che avviene nella pila è come sappiamo la seguente
Cu2+ + Zn
 Cu + Zn2+
Cu + Zn2+Quando tale reazione avrà raggiunto l'equilibrio non vi sarà più trasferimento di elettroni, la pila sarà esaurita per cui la differenza di potenziale sarà pari a zero.
Possiamo dunque affermare che quando le specie chimiche avranno raggiunto le loro concentrazioni di equilibrio E = 0.All'equilibrio varrà allora

Ma il rapporto di cui si calcola il logaritmo non è altro che la costante di equilibrio della reazione kc.
Potremo perciò scrivere
relazione tra forza elettromotrice di una pila in condizioni standard e costante di equilibrio della reazione redox alla base della pila stessa.
Si tenga presente che misurare sperimentalmente i potenziali risulta molto spesso più semplice che misurare le concentrazioni di equilibrio.
In tal modo mediante la misura della f.e.m. in condizioni standard (o più semplicemente mediante la consultazione dei potenziali standard già tabulati) è possibile calcolare la kc di molte reazioni redox.Lavoro eseguito da una pila
Ricordando che la differenza di potenziale elettrico è data dal rapporto tra lavoro e carica elettrica (V = L/Q) ed il volt (unità di misura della differenza di potenziale elettrico) si esprime in Joule su Coulomb (J/C), sarà allora possibile calcolare il lavoro eseguito da una pila come
L = V . Q
Poiché il voltaggio si riferisce al singolo elettrone, per calcolare la carica elettrica Q complessivamente trasportata durante il lavoro della pila quando tutte le concentrazioni sono 1 M, sarà necessario moltiplicare il numero n di elettroni effettivamente scambiati per la carica portata da 1 mole di elettroni (1 Faraday)
Q = n . F
In condizioni standard infine
V = DE°
per cui il lavoro compiuto da una pila in condizioni standard sarà pari a
L = DE° . n . F
ad esempio la pila Daniell, che presenta DE° = 1,1 V, è in grado di fornire
L = 1,1 V . 2 . 96485 C = 212,3 KJ/mol
Elettrolisi e celle elettrolitiche
Come abbiamo già avuto modo di dire l'elettrolisi è un processo in cui energia elettrica viene impiegata per far avvenire una reazione redox che si produrrebbe spontaneamente in senso opposto.
Se immergiamo due elettrodi collegati ad un generatore di corrente continua in un recipiente che contenga NaCl fuso (e quindi dissociato in ioni Na+ e Cl-), gli ioni Na+ vengono attratti dall'elettrodo negativo dove si riducono acquistando un elettrone, mentre gli ioni Cl- vengono attratti dall'elettrodo positivo al quale cedono il loro elettrone ossidandosi.
Poiché in questo caso la reazione di riduzione avviene all'elettrodo negativo questo prende il nome di catodo, mentre l'elettrodo positivo, dove avviene l'ossidazione, è l'anodo.
Si noti che i segni risultano opposti rispetto ad una pila.

Dunque al catodo gli ioni Na+ si riducono a sodio metallico, mentre all'anodo gli ioni Cl- si ossidano a Cloro gassoso Cl2.
Gli elettroni fluiscono quindi dal catodo negativo all'anodo positivo in modo analogo a quanto avverrebbe se collegassimo i due elettrodi mediante un filo metallico.
La conduzione elettrica attraverso un filo metallico è detta conduzione di prima specie, mentre la conduzione che avviene grazie alle reazioni degli ioni presenti nel fluido è detta conduzione elettrolitica o di seconda specie.
Nel caso della conduzione elettrolitica gli elettroni che abbandonano il catodo non sono gli stessi che entrano nell'anodo, ma l'effetto complessivo è identico. Attraverso l'uso di un amperometro possiamo infatti verificare la presenza di una corrente elettrica che attraversa il circuito, sia che esso venga chiuso mediante un conduttore metallico, sia che venga chiuso attraverso una soluzione di elettroliti.La reazione complessiva che avviene in soluzione è la seguente

In assenza della corrente elettrica fornita dall'esterno tale reazione sarebbe naturalmente spontanea in senso contrario.
Possiamo dunque definire l'elettrolisi un processo di decomposizione di un composto, fuso o in soluzione, in cui il passaggio di corrente elettrica produce una reazione redox endoergonica.
Naturalmente solo i composti che si dissociano in ioni possono subire l'elettrolisi e per tale motivo essi sono comunemente detti elettroliti.
Elettrolisi di una soluzione contenente più ioni (precedenza di scarica)
In una soluzione acquosa di un elettrolita la situazione viene complicata dalla presenza in soluzione di ioni H+ e ioni OH- provenienti dalla parziale dissociazione dell'acqua.
Ad esempio se effettuiamo l'elettrolisi di una soluzione acquosa di cloruro di sodio, possiamo prevedere che all'anodo competeranno per la reazione di ossidazione sia gli ioni Cl- che gli ioni OH-, mentre al catodo si presenteranno gli ioni Na+ e gli ioni H+.Il problema in questo caso è prevedere quale delle specie ioniche presenti in soluzione, e che competono per uno stesso elettrodo, ha la precedenza di scarica.
Si verifica sperimentalmente che, a parità di concentrazione, la precedenza di scarica dipende essenzialmente dalla maggiore o minore tendenza ad acquistare o a cedere elettroni, tendenza che viene misurata attraverso i potenziali standard di riduzione.
Precedenza di scarica al catodo
Tenendo presente che al catodo le specie accettano elettroni e si riducono, è logico attendersi che si ridurrà per prima la specie che presenta il potenziale standard di riduzione più elevato.Precedenza di scarica all'anodo
Tenendo presente che all'anodo le specie cedono elettroni e si ossidano, è logico attendersi che si ossiderà per prima la specie che presenta il potenziale standard di riduzione più basso.Nel caso in cui le specie siano presenti in concentrazione non standard è necessario confrontare i potenziali non standard calcolati con l'equazione di Nernst.
Sulla base di tali considerazioni proviamo dunque a prevedere quel che succede in una soluzione acquosa di Ioduro di potassio (KI) 1M.
1) all'anodo competono per la reazione di ossidazione gli ioni OH-, I- e H2O. Le reazioni di riduzione ed i relativi potenziali standard sono
 )
)
 )
)
 )
) Se tutte le specie chimiche fossero presenti in concentrazione 1M è evidente che si ossiderebbero per primi gli ioni OH- che presentano un potenziale di riduzione minore con liberazione di ossigeno gassoso all'anodo.
Dobbiamo però tener presente che in una soluzione neutra la concentrazione degli ioni OH- e degli ioni H+ non è 1 M, ma 10-7 M e la pressione parziale dell'ossigeno atmosferico è circa di 0,2 atmosfere. Calcoliamo quindi il potenziale delle coppie redox O2/OH- e H2O/O2 a pH 7, utilizzando l'equazione di Nernst
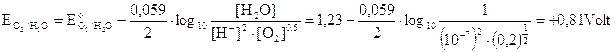
Come si può osservare a pH 7 il potenziale più basso risulta essere in realtà quello dello iodio (0,54 contro 0,81) che si scarica dunque per primo all'anodo liberando I2 che colora di rosso la soluzione nei pressi dell'elettrodo.
2) Al catodo competono per la scarica gli ioni H+, K+ e l'acqua. Le reazioni di riduzione ed i relativi potenziali standard sono
 )
) )
) )
)Anche in questo caso, se tutte le specie chimiche fossero presenti in concentrazione 1M è evidente che si ridurrebbero per primi gli ioni H+ che presentano un potenziale di riduzione di gran lunga maggiore.
Tenendo però presente che in una soluzione neutra la concentrazione degli ioni H+ e degli ioni OH- non è 1 M, ma 10-7 M, dobbiamo applicare l'equazione di Nernst. A pH 7 i potenziali di riduzione assumono i seguenti valori

Nonostante la variazione dei potenziali, la liberazione di idrogeno gassoso al catodo rimane la reazione favorita.
Poiché nella soluzione rimangono ioni K+ e ioni OH-, il processo elettrolitico ha prodotto una soluzione di idrossido di potassio.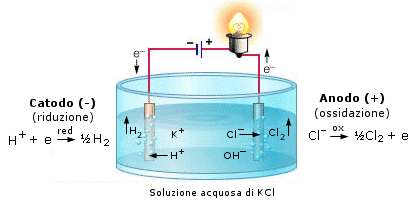
ALTRI ESEMPI
La soda caustica
In modo analogo l'elettrolisi di una soluzione acquosa di cloruro di sodio, liberando cloro gassoso all'anodo (Cl2) ed idrogeno gassoso al catodo (H2) è in grado di arricchire la soluzione di idrossido di sodio (soda caustica NaOH). L'elettrolisi del cloruro di sodio in soluzione acquosa è il processo industriale per la produzione della soda caustica. Durante la produzione è necessario tener separato il compartimento anodico da quello catodico per evitare che il cloro nascente dismuti, a contatto con l'idrossido di sodio, ridando cloruro ed ipoclorito (varechina)2NaOH + Cl2 → NaCl +NaClO + H2O
L'acido solforico
Si può ottenere eseguendo l'elettrolisi di un solfato di un metallo il cui potenziale di riduzione sia sufficientemente elevato (maggiore di ).
).Si tenga presente che in una soluzione di solfato rameico il calcolo dei potenziali tramite l'equazione di Nernst deve tener conto del fatto che tale sale da idrolisi acida e quindi il pH 7.
Gli ioni rameici Cu2+ in acqua formano un complesso di coordinazione con 6 molecole d'acqua (ione esaacquo rameico), il quale si comporta come un acido debole dissociandosi secondo la reazioneCu(H2O)62+ → [Cu(H2O)5(OH)]+ + H+
in uno ione pentaacquo-monossi-rameico e uno ione H+. La costante di dissociazione acida dello ione rameico esacoordinato è pari a ka =1,58.10-7.
La concentrazione di ioni H+ di una soluzione 1 M di solfato rameico sarà pertanto
ed il pH = - log 4.10-4 = 3,4
A pH 3,4 i potenziali delle coppie redox che competono per la scarica al catodo diventano


si riduce quindi il rame che presenta un potenziale superiore.
Sempre a pH 3,4 il potenziale delle coppie redox che competono per la scarica all'anodo diventa
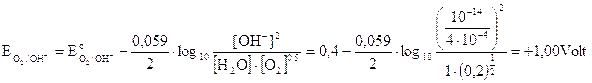

Nonostante il potenziale sia ulteriormente salito all'anodo continua a liberarsi ossigeno in quanto il potenziale di riduzione dell'anione solfato rimane di gran lunga più elevato.
Elettrolisi dell'acqua
Se al posto del solfato rameico tentiamo di effettuare l'elettrolisi di un solfato di un metallo il cui potenziale di riduzione sia meno elevato rispetto a quello delle altre coppie redox che competono per la riduzione (ad esempio solfato di sodio ) oppure di una soluzione diluita di acido solforico, allora al catodo si svilupperà idrogeno, mentre all'anodo continuerà a svilupparsi ossigeno.
) oppure di una soluzione diluita di acido solforico, allora al catodo si svilupperà idrogeno, mentre all'anodo continuerà a svilupparsi ossigeno.
Si sarà in tal modo ottenuta l'elettrolisi dell'acqua2H2O → 2H2 + O2
Si tenga presente che il fenomeno risulta sensibile solo se in soluzione esiste un elettrolita che consenta la conduzione. Se tentiamo di effettuare l'elettrolisi dell'acqua pura il passaggio di corrente sarà praticamente nullo a causa della bassissima costante di dissociazione dell'acqua, ed il processo elettrolitico assolutamente trascurabile. Si noti infine come per ogni molecola di ossigeno se ne liberino due di idrogeno e dunque l’idrogeno liberato occupa un volume doppio dell’ossigeno.

Le leggi di Faraday
Gli aspetti quantitativi dell'elettrolisi furono studiati da Faraday, il quale sintetizzò i risultati ottenuti sperimentalmente in due leggi.
Prima legge di Faraday
La massa di una sostanza che si libera agli elettrodi è proporzionale alla quantità di elettricità passata attraverso la soluzione.Dopo quanto osservato a proposito dei fenomeni elettrolitici tale legge dovrebbe risultare abbastanza evidente. Se ad esempio riprendiamo in considerazione l'elettrolisi del cloruro di sodio fuso, possiamo verificare che per ogni elettrone che abbandona il catodo e raggiunge l'anodo passando attraverso la soluzione, si libera 1 atomo di Na metallico e mezza molecola di Cloro gassoso

Naturalmente se passano 2 elettroni si libereranno 2 atomi di sodio e 1 molecola di cloro ed in generale se passano un numero n di elettroni si libereranno agli elettrodi n atomi di sodio e n/2 molecole di cloro.
Risulta allora evidente come la quantità di materia che si libera agli elettrodi sia proporzionale al numero di elettroni scambiati. Ma poiché ogni elettrone porta una quantità di elettricità costante e definita (1,6.10-19 C) se ne deduce che la massa liberata deve essere proporzionale alla carica elettrica transitata attraverso la soluzione.Seconda legge di Faraday
Al fine di liberare agli elettrodi 1 equivalente di una qualsiasi sostanza è necessario che attraverso la soluzione passino 96.485 C (quantità nota come Faraday). Naturalmente se attraverso la soluzione passano n Faraday verranno liberati n equivalenti.Il Faraday (F) è la quantità di elettricità portata da 1 mole di elettroni ( N elettroni)
N . Carica elettrone = 6,0221415.1023 . 1,60217653.10-19 = 96.485,336 C
Ricordando che il peso equivalente di una sostanza che subisce un processo redox è pari al rapporto tra il suo peso molare ed il numero di elettroni persi o acquistati

risulta evidente come tale quantità sia in grado di acquistare o cedere in un processo redox esattamente una mole di elettroni, pari alla carica di 96.485 C (1 Faraday).Ad esempio se vogliamo ridurre 1 mole di Al3+ (PM = 27 g) ad alluminio metallico
Al3+ + 3e- → Al
poiché ogni ione Al3+ richiede 3 elettroni, 1 mole (27 g), contenendo N ioni, richiede 3N elettroni.
N elettroni riusciranno a ridurre solo un terzo di mole, cioè 1 equivalente di alluminio (9 g).Infine se 1 Faraday libera 1 equivalente è evidente che n Faraday libereranno n equivalenti, dal che si deduce che la massa che si libera agli elettrodi in una reazione redox sta al peso equivalente come la quantità di corrente che attraversa la soluzione sta a 96.485 C.
Per calcolare la massa che si libera ad un elettrodo sarà dunque sufficiente moltiplicare il peso equivalente per il numero di Faraday che hanno attraversato la soluzione

ESEMPIO
Calcoliamo quanto rame metallico si deposita al catodo di una cella elettrolitica contenente CuSO4, quando si faccia passare una corrente di 15 A per 2,5 ore.Sapendo che 1 A = 1 C/s, la quantità totale di corrente che attraversa la soluzione è pari a
Q = I . t = 15 C/s . 9000 s = 135.000 C
sapendo che 1 Faraday = 96.485 C, allora 135.000C corrispondono a

Calcoliamo ora il peso equivalente del rame

Dunque se 1 Faraday libera 1 equivalente di rame, 1,4 Faraday ridurranno 1,4 equivalenti pari a
31,77 . 1,4 = 44,478 g
Equivalente elettrochimico
Si definisce equivalente elettrochimico il rapporto tra il peso equivalente e la carica elettrica portata da 1 Faraday.
equivalente elettrochimico =
L'equivalente elettrochimico rappresenta quindi la quantità in grammi di una sostanza liberata al passaggio di 1 Coulomb.
Ad esempio l'equivalente elettrochimico dell'alluminio è pari a
Fenomeni elettrochimici di interesse pratico
1) La corrosione
Qualsiasi elemento che presenti un potenziale di riduzione inferiore a quello della coppia H2O/H2,OH- può essere ossidato dall'acqua, la quale ne acquista gli elettroni riducendosi secondo la reazione )
)Naturalmente il potenziale di tale reazione è misurato in condizioni standard, per una concentrazione 1 M di ioni OH- (pH = 14). Calcolando il potenziale di tale reazione per un pH 7, tramite l'equazione di Nernst, si ottiene
a pH = 7
L'acqua pura a pH neutro è quindi in grado di ossidare (corrodere) tutti i metalli che presentano un potenziale inferiore a -0,42V come lo zinco ( -0,76) l'alluminio (-1,66) e tutti i metalli alcalini e alcalino-terrosi.
Il ferro, , possedendo un potenziale praticamente uguale a quello dell'acqua non viene praticamente attaccato a pH = 7 ( le soluzioni acide però lo corrodono).
, possedendo un potenziale praticamente uguale a quello dell'acqua non viene praticamente attaccato a pH = 7 ( le soluzioni acide però lo corrodono).Tuttavia se l'acqua contiene ossigeno o è presente umidità nell'aria è necessario tener presente la seguente semireazione di riduzione
 )
)a pH = 7 la coppia H2O,O2/OH- presenta potenziale pari a 0,81 V ed è quindi in grado di ossidare non solo il ferro, ma molti altri metalli come Stagno (- 0,14 V), Piombo (- 0,13 V) e Rame (+ 0,34 V).
Si tenga inoltre presente che in molti testi viene considerata la seguente semireazione di riduzione
 )
)
che a pH = 7 presenta presenta il medesimo potenziale pari a 0,81 V ed è dunque perfettamente equivalente precedente alla precedente (la prima libera infatti 4 ioni OH-, mentre la seconda consuma 4 ioni H+)La corrosione del ferro ad opera dell'acqua contenente ossigeno atmosferico (arrugginimento) è un fenomeno complesso e non completamente chiarito. Si ritiene che il ferro metallico venga dapprima ossidato a Fe2+ dalla coppia H2O,O2/OH-.
2Fe + O2 + 2H2O → 2Fe(OH)2
Gli ioni ferrosi che passano in soluzione vengono rapidamente ossidati a ioni ferrici quando giungono a contatto con regioni acquose a maggior tenore di ossigeno.
4Fe(OH)2 + O2 + 2H2O → 4Fe(OH)3
Sommando membro a membro si ottiene
4Fe + 3O2 + 6H2O → 4Fe(OH)2
Non appena formatisi gli ioni ferrici producono idrossidi e carbonati insolubili (grazie alla CO2 atmosferica che si scioglie in acqua formando acido carbonico), che precipitano in un solido scaglioso di composizione non perfettamente definita noto come ruggine.
In realtà si ritiene più probabile che l'idrossido ferrico precipiti sotto forma di ossido ferrico idratato, attraverso una reazione del tipo
2Fe(OH)3 → Fe2O3.3H2O
Nella ruggine l'ossido ferrico sarebbe presente con diverso grado di idratazione, pertanto si preferisce descriverlo con la formula generale Fe2O3.nH2O.

2) La passivazione
Molti altri metalli (Zinco, Rame, Alluminio etc) reagiscono all'aria umida, ma a differenza di quanto accade per il ferro, la miscela di ossidi e carbonati che si forma non è friabile, ma costituisce uno strato solido, coerente, che protegge il metallo sottostante da ulteriori fenomeni corrosivi. Tale processo è detto passivazione.Ad esempio il rame (
 ) all'aria umida viene ossidato con formazione di idrossido rameico il quale reagisce in parte con la CO2 disciolta in acqua per dare un carbonato monobasico rameico, responsabile della patina verde che ricopre il metallo esposto all'aria (verderame)
) all'aria umida viene ossidato con formazione di idrossido rameico il quale reagisce in parte con la CO2 disciolta in acqua per dare un carbonato monobasico rameico, responsabile della patina verde che ricopre il metallo esposto all'aria (verderame)Cu(OH)2 + H2CO3 [Cu(OH)]2CO3 + 2 H2O
Lo zinco si comporta in modo analogo formando sulla sua superficie uno strato passivante costituito da ossido ZnO e carbonato basico [Zn(OH)]2CO3.
La passivazione dell'alluminio può essere addirittura effettuata artificialmente, ottenendo uno strato di ossido più spesso, utilizzando il metallo come anodo in una cella elettrolitica. In tal modo lo strato superficiale del metallo si ossida e si ottiene l'alluminio anodizzato.
3) Prevenzione della corrosione
Per proteggere il ferro dalla corrosione (si calcola che circa il 20% della produzione annua mondiale di ferro venga utilizzata per sostituire il metallo arrugginito) è necessario impedire che la coppia H20,O2/OH- sia in grado di esercitare i suoi effetti.Una soluzione semplice è quella di verniciare il ferro con una vernice antiruggine contenente sostanze chimiche, come il minio Pb3O4, il quale, avendo un potenziale normale di +1,45 Volt, non può essere ossidato dall'aria umida.
Naturalmente le vernici antiruggine rappresentano una soluzione per piccole strutture metalliche e comunque di non lunga durata, non essendo in grado di aderire permanentemente alla superficie metallica.
Una soluzione più duratura è quella di rivestire il ferro con uno strato sottile di un metallo con potenziale di riduzione più basso del ferro stesso, ma in grado di subire il fenomeno della passivazione come ad esempio lo zinco.
Il ferro zincato viene egregiamente protetto dalla corrosione, sia perché in superficie si forma uno strato inerte di ossidi e carbonati di zinco, sia perché, anche se lo strato di zinco viene scalfito ed il ferro esposto agli agenti atmosferici, lo zinco ne previene ulteriormente la corrosione ossidandosi al posto del ferro (presenta infatti un potenziale minore).
La zincatura può essere effettuata immergendo il ferro nello zinco fuso (automobili) o usando l'oggetto da zincare come catodo in una cella elettrolitica contenente un sale di zinco (galvanizzazione)Non così accadeva con il ferro stagnato (latta) in quanto lo stagno presenta un potenziale di riduzione (-0,14 V) più elevato del ferro. Una volta scalfita la superficie stagnata il ferro arrugginisce.
Per lo stesso motivo non si dovrebbero fissare oggetti di rame (ad esempio grondaie) con chiodi di ferro, in quanto il rame presenta un potenziale superiore al ferro e tende quindi ad ossidarlo rapidamente (in presenza di una soluzione elettrolitica come può essere l'umidità atmosferica che si condensa sulle superfici metalliche).
Naturalmente non è possibile zincare strutture metalliche di grandi dimensioni come navi o ponti. In tal caso è sufficiente collegare la struttura ad una placca di metallo a potenziale minore, detto anodo sacrificale (in genere Zinco o Magnesio) che si ossida al posto del ferro. Naturalmente tali placche vanno periodicamente sostituite.4) Galvanoplastica
Il processo di galvanizzazione, che abbiamo visto essere utilizzato per zincare e proteggere il ferro dalla corrosione, può essere utilizzato anche per ricoprire (placcare) oggetti anche con metalli diversi, sia per protezione che a scopi decorativi.
Se ad esempio vogliamo argentare un oggetto di ferro è sufficiente utilizzarlo come catodo in una cella elettrolitica contenente un sale di argento (ad esempio AgNO3), mentre all'anodo useremo un elettrodo di argento metallico. In tal modo l'argento metallico si ossida passando in soluzione come Ag+, mentre al catodo gli ioni Ag+ si depositano come argento metallico.
In modo analogo si possono eseguire altre placcature elettrolitiche come dorature, nichelature, cromature etc.
Quando l'oggetto da rivestire non è un conduttore lo si può rendere tale ricoprendolo con una vernice conduttrice (ad esempio polvere di grafite).
Quando l'operazione viene eseguita rendendo conduttrice la parte interna di uno stampo è possibile far depositare il metallo all'interno dello stesso ottenendo in tal modo oggetti metalli per via elettrolitica.6) Raffinazione elettrolitica dei metalli
Con un processo analogo alla placcatura elettrolitica è possibile procedere alla raffinazione dei metalli. Se infatti in una soluzione elettrolitica che contenga un sale del metallo da raffinare, usiamo come anodo il metallo grezzo da raffinare e come catodo una lamina dello stesso metallo allo stato puro, il processo elettrolitico porterà in soluzione l'anodo ossidandolo, mentre depositerà al catodo metallo puro riducendo gli ioni metallici in soluzione.Ad esempio per la raffinazione elettrolitica del rame si immergono gli elettrodi di rame (l'anodo impuro, il catodo puro) in una soluzione di solfato rameico e acido solforico.

Elementi di termodinamica chimica
Per descrivere in modo completo una reazione chimica non è sufficiente specificare il numero di moli di reagenti e prodotti di reazione che vi partecipano, ma è necessario descrivere anche i fenomeni energetici che l'accompagnano.
Tale descrizione è compito della termodinamica la quale permette di stabilire se una reazione chimica, scritta sulla carta, avvenga effettivamente in pratica.
In definitiva, attraverso lo studio delle trasformazioni energetiche che accompagnano sempre i fenomeni chimici, è possibile evidenziare i criteri di spontaneità delle reazioni e definire quali sono le condizioni termodinamiche affinché una reazione possa prodursi con la massima efficienza.E' bene comunque ricordare che la termodinamica, occupandosi dei sistemi al punto di equilibrio, non è in grado di stabilire con che velocità una reazione avviene, ma solo se si produce e in che proporzione.
I sistemi termodinamici
Il termine sistema viene qui impiegato per descrivere la regione di spazio cui vengono limitate le nostre osservazioni. Tutto ciò che non appartiene al sistema è detto ambiente esterno o intorno del sistema. Il sistema più il suo intorno costituiscono complessivamente l’universo.
In linea di principio l’universo è costituito dall’universo propriamente detto, ma in pratica con questo termine si indica solo il sistema e quella parte dell’ambiente esterno con cui il sistema è a diretto contatto. Un sistema chimico è tipicamente composto dalle specie chimiche coinvolte nella reazione, mentre possiamo in pratica limitare l'ambiente esterno al recipiente che le contiene ed alla porzione di spazio circostante con cui il sistema intrattiene eventuali scambi di energia e/o di materia.

a) Si definiscono aperti i sistemi in grado di scambiare energia e materia con l'ambiente
b) Si definiscono chiusi i sistemi in grado di scambiare solo energia con l'ambiente
c) Si definiscono isolati i sistemi che non sono in grado di effettuare scambi con l'ambiente.

Come tutti i sistemi anche un sistema chimico possiede energia.
L'energia viene definita come la capacità di compiere lavoro o di trasferire calore. Le unità di misura dell'energia sono quindi le stesse del lavoro e del calore.
Il calore (q) è l’energia termica trasferita tra il sistema e l’ambiente come risultato di differenze di temperatura.
Il lavoro (w) è l’energia trasferita tra il sistema e l’ambiente come risultato di una forza che opera su una distanza.
lavoro = forza x distanza
Le unità di misura maggiormente utilizzate in chimica sono il joule ( 1 J = Newton . metro = 1 kg . m2 . s-2) e la caloria (1 cal = energia necessaria per aumentare di 1°C la temperatura di 1 g d'acqua)1 cal = 4,184 J
Funzioni di stato e di percorso
Il valore assunto da una funzione di stato dipende solamente dalle condizioni in cui si trova il sistema. È una caratteristica del sistema ed è indipendente dal modo in cui il sistema sia giunto in quelle condizioni. Sono funzioni di stato la temperatura, il volume, l'energia.
Il valore assunto da una funzione di percorso dipende dal percorso effettuatoedè una caratteristica del processo in esame. Sono funzioni di percorso il lavoro e il calore.
Ad esempio, l’altezza di una montagna è indipendente dal tragitto effettuato per arrivarci (l’altezza è una funzione di stato), mentre la lunghezza della strada dipende dal tragitto scelto per arrivare in cima (la lunghezza della strada e una funzione di percorso).
Energia interna (E)
L'energia posseduta da un sistema viene definita Energia interna E (spesso indicata anche come U).
L'energia interna di un sistema dipende esclusivamente dalla pressione, dalla temperatura, dal volume e dalla composizione chimica del sistema (tipo ed intensità delle interazioni tra particelle costituenti).
Per questo motivo l'energia interna è una funzione di stato. Il suo valore dipende cioè dallo stato termodinamico del sistema e non dal modo in cui tale condizione è stata raggiunta.
L'energia interna di 100 g di acqua a 25°c e 1 atm è la medesima che l'acqua sia stata ottenuta per condensazione di 100 g di vapore, per reazione tra idrogeno ed ossigeno o per liquefazione di 100 g di ghiaccio.Inoltre l'energia interna è una proprietà estensiva, è cioè proporzionale alla quantità di materia presente nel sistema.
100 g di acqua a 25°C e 1 atm possiedono un'energia interna doppia rispetto a 50 g di acqua nelle stesse condizioni termodinamiche.L'energia interna di un sistema chimico è uguale alla somma dell'energia cinetica e dell'energia potenziale di tutte le molecole che formano il sistema.ed è associata ai moti traslatori, rotazionali, vibrazionali, elettronici e nucleari delle molecole, nonché alle interazioni interatomiche (legami intramolecolari) ed intermolecolari.
E = (Ec + Ep)
L’energia interna non include invece l'energia cinetica e potenziale associata al sistema nel suo complesso, ad esempio, se il sistema è in movimento o si trova in un campo gravitazionale o elettromagnetico.
L'energia cinetica di un corpo (Ec) è la capacità di compiere lavoro per effetto del suo moto. Essa dipende dalla massa m e dalla velocità v ed è pari a
Come abbiamo già anticipato, nel caso di un sistema di molecole, l'energia cinetica non è legata solo al movimento di traslazione, ma vi sono altri contributi connessi con i moti di rotazione e di vibrazione interna delle singole particelle.

La meccanica statistica dimostra che il valore medio dell'energia cinetica di un sistema di particelle è legato alla temperatura del sistema dalla relazione

E dunque l’energia cinetica di un sistema rappresenta la sua energia termica.L'energia potenziale di un corpo è la sua capacità di compiere lavoro per effetto della sua posizione o dello stato in cui si trova.
Nel caso di un sistema di particelle l'energia potenziale viene comunemente definita energia chimica o di legame e presenta due componenti fondamentali. Una legata alla posizione reciproca delle molecole il cui valore dipende dalle forze attrattive intermolecolari (forze di Van der Waals), l'altra che dipende dalla posizione reciproca assunta dai protoni e dagli elettroni all'interno degli atomi e dalle interazioni elettromagnetiche che si producono tra le loro cariche e dunque dai legami chimici interatomici (covalente, ionico e metallico).
Quando effettuiamo un lavoro per allontanare due corpi che si attraggono (come accade tra molecole o tra elettroni e nuclei) allora l'energia potenziale del sistema aumenta e le forze di attrazione tra i corpi diminuiscono.

Al contrario quando lasciamo che due corpi che si attraggono si avvicinino l'energia potenziale diminuisce e le forze di attrazione tra i corpi aumentano.

Le reazioni chimiche sono sempre accompagnate da modificazioni energetiche in cui l'energia potenziale varia a causa delle modificazioni delle posizioni reciproche di atomi e di elettroni che si riassestano per occupare nuove posizioni nelle molecole dei prodotti di reazione.
In altre parole ogni reazione chimica avviene grazie alla rottura ed alla formazione di legami che modificano l'intensità delle forze all'interno delle molecole e tra le molecole, andando a modificare l'energia potenziale del sistema.Se i nuovi legami che si formano sono più deboli il sistema presenta un'energia potenziale maggiore rispetto a quella che caratterizzava nel complesso le molecole dei reagenti.
Se al contrario i nuovi legami che si formano sono più forti il sistema presenta un'energia potenziale minore rispetto a quella che caratterizzava nel complesso le molecole dei reagenti.Se il sistema è isolato e non può quindi scambiare energia con l'ambiente esterno la reazione avviene grazie alla conversione di parte dell'energia interna da cinetica in potenziale o viceversa (trasformazione adiabatica).
In altre parole una reazione in un sistema isolato trasforma sempre parte dell'energia cinetica delle molecole (energia termica) in energia potenziale (energia chimica di legame) ed il sistema si raffredda, o, viceversa, trasforma parte dell'energia di legame in energia termica ed il sistema si riscalda.Tale affermazione è una conseguenza del primo principio della termodinamica che afferma che l'energia non si può né distruggere né creare, ma può solo essere convertita da una forma in un'altra.
Se invece il sistema può scambiare energia con l'intorno, il primo principio della termodinamica ci informa che la quantità di energia che il sistema scambia con l'ambiente esterno è esattamente pari alla sua variazione di energia interna.
In altre parole l'energia totale dell'universo (sistema + ambiente esterno) rimane sempre costante.
In realtà non è possibile calcolare l'energia interna di un sistema, ma solo le variazioni di energia che caratterizzano due stati diversi di un sistema (ciò è dovuto al fatto che parte dell'energia interna di un sistema è costituita da energia potenziale di cui, come sappiamo, non è possibile determinare un valore assoluto).Se ad esempio un sistema chimico reagisce trasformando dei reagenti in prodotti di reazione, noi siamo in grado di misurare solo la variazione di energia interna E che si verifica durante la reazione, espressa come differenza tra l'energia interna dei prodotti di reazione (energia interna dello stato finale, Ef) e l'energia interna dei reagenti (energia interna dello stato iniziale, Ei)
E = Ef − Ei
Non ci è invece possibile misurare i valori assoluti di Ef ed Ei.
In un sistema isolato E = 0 e la reazione avviene per conversione interna di una forma di energia in un'altra (cinetica in potenziale, o viceversa).
Nella maggior parte delle reazioni chimiche il sistema è in grado di scambiare energia con l'ambiente, per cui E 0.
Variazioni dell'energia interna E di un sistema chimico
Durante una reazione chimica l'energia interna di un sistema può variare essenzialmente poiché avvengono scambi di calore q e/o di lavoro w con l'ambiente esterno.
Determinare la variazione . E di energia interna associata ad una reazione chimica significa misurare l’entità del calore e del lavoro scambiati dal sistema con l’ambiente.Si tenga presente che i flussi di energia in uscita dal sistema, diminuendone l’energia interna, assumono valore negativo, mentre i flussi di energia in entrata nel sistema, aumentandone l’energia interna, assumono valore positivo.

Scambi di calore
Quando un sistema assorbe calore la sua energia interna aumenta, mentre quando un sistema cede calore la sua energia interna diminuisce. Per questo motivo al calore assorbito dal sistema viene assegnato valore positivo, mentre al calore ceduto viene assegnato valore negativo.A + B + q (calore) → C + D reazione endotermica
A + B − q (calore) → C + D reazione esotermica
La reazione esotermica può ovviamente essere scritta anche riportando il calore di reazione con il segno positivo tra i prodotti di reazione
A + B → C + D + q (calore) reazione esotermica
Supponiamo ora per semplicità che una reazione avvenga solo con scambio di calore tra il sistema e l'ambiente. Potremo allora scrivere
E = q
In assenza di lavoro, lo scambio di calore è uguale alla variazione dell'energia interna del sistema
In pratica il calore scambiato in una reazione isocora (e in assenza di qualsiasi altro lavoro oltre a quello di espansione) può essere utilizzato come misura della variazione di energia interna di un sistema.Prendiamo ad esempio la reazione di combustione del glucosio a 25°C ed 1 atm
C6H12O6(s) + 6O2(g) → 6CO2(g) + 6H2O(l) + 2808 kJ/mol
che possiamo anche scrivere
C6H12O6(g) + 6O2(g) - 2808 kJ/mol → 6CO2(g) + 6H2O(l)
a testimonianza del fatto che l'energia interna del sistema è diminuita di 2808 kJ per ogni mole di glucosio bruciato. Tale energia è stata completamente ceduta all'ambiente sotto forma di calore.
Per tale reazione possiamo scrivereE = q = − 2808 kJ/mol
In questo esempio il sistema ha scambiato solo calore con l'ambiente e per questo motivo la variazione di energia interna coincide con il calore scambiato, ma in generale durante le razioni chimiche un sistema può eseguire un lavoro sull'ambiente o, viceversa, l'ambiente può effettuare un lavoro sul sistema.
Scambi di lavoro
E' evidente che in questo caso l'energia interna del sistema varierà di conseguenza. Se il sistema esegue un lavoro sull'ambiente lo fa a spese della sua energia interna che diminuisce, mentre se l'ambiente effettua un lavoro sul sistema l'energia interna di quest'ultimo deve aumentare.
Per questo motivo al lavoro eseguito dal sistema viene assegnato valore negativo, mentre al lavoro subito dal sistema viene assegnato valore positivo.Se teniamo quindi conto sia degli scambi di calore che del lavoro compiuto, la variazione di energia interna assume la forma
E = q + w
Tale relazione esprime in modo completo il primo principio della termodinamica, affermando che la variazione di energia interna di un sistema dipende dal calore scambiato con l'ambiente e dal lavoro eseguito durante la trasformazione.
Nella maggior parte delle reazioni chimiche il lavoro prodotto durante le trasformazioni è legato alle variazioni di volume del sistema in seguito ad un cambiamento nel numero totale delle moli delle specie chimiche gassose. In questo caso il sistema è libero di espandersi (se il numero di moli aumenta) eseguendo un lavoro sull’ambiente contro la pressione esterna (in genere la pressione atmosferica) o di contrarsi (se il numero di moli diminuisce) subendo un lavoro da parte dell’ambiente.
Ora è semplice dimostrare che il lavoro di espansione eseguito da un gas contro una pressione esterna costante P è pari a
wesp = − P . Vmentre il lavoro di contrazione subito da un gas sottoposto alla pressione esterna costante P è pari a
wcont = + P . V
dove V è la variazione di volume
Infatti il lavoro è dato dal prodotto di una forza F per lo spostamento x generato dalla medesimaw = F . x
ricordando tuttavia che la Pressione è il rapporto tra una forza F e la superficie di area A sulla quale essa agisce P = F/A e quindi F = P.A, potremo scrivere w = P . A . x = P . V
Ricordando infine che la variazione di volume V è pari a

il lavoro compiuto sarà allora pari a

Usando per la costante dei gas il valore R = 8,314
 , si ottiene direttamente il lavoro espresso in joule.
, si ottiene direttamente il lavoro espresso in joule.Il sistema compie lavoro ( w negativo, l'energia interna diminuisce)
Se durante la reazione il numero complessivo delle moli gassose aumenta il sistema si espande utilizzando parte della sua energia interna per eseguire un lavoro, in genere contro l'atmosfera sovrastante. Il lavoro eseguito va a diminuire l'energia interna del sistema.Prendiamo ad esempio in considerazione la reazione a 25°C e 1 atm
Zn(s) + 2HCl(aq) → ZnCl2(aq) + H2(g) + 153,89 kJ
La reazione libera 153,89 kJ sotto forma di calore ceduto all'ambiente, per cui l'energia interna del sistema diminuirà di conseguenza e potremo scrivere
q = − 153,89 kJ
Durante la reazione si forma però una mole di idrogeno gassoso. Il sistema si espande compiendo un lavoro contro il peso dell'atmosfera sovrastante.

Calcoliamo allora il lavoro eseguito dal sistema durante l'espansione di 1 mole di idrogeno a 25°C e 1 atm

Possiamo allora affermare che non tutta l'energia ceduta dal sistema è stata dissipata come calore. Parte di essa è stata utilizzata dal sistema per eseguire un lavoro sull'ambiente pari a 2,48 kJ. Per questo motivo l'energia interna del sistema dovrà diminuirà dello stesso valore e di conseguenza potremo scrivere
w = − 2,48 kJ
La variazione complessiva dell'energia interna, tenendo conto sia del calore ceduto che del lavoro eseguito sarà allora pari aE = q + w = ( − 153,89 kJ ) + ( − 2,48 kJ ) = − 156,37 kJ
La diminuzione di energia interna risulta quindi in tal caso leggermente superiore alla cessione di calore, a causa del lavoro di espansione del sistema.
L'ambiente compie lavoro sul sistema ( w positivo, l'energia interna aumenta)
Se durante la reazione il numero complessivo delle moli gassose diminuisce il sistema si contrae, subendo un lavoro di compressione, in genere da parte dell'atmosfera sovrastante. Il lavoro eseguito dall'ambiente va ad aumentare l'energia interna del sistema.Prendiamo ad esempio in considerazione la reazione a 25°C e 1 atm
3H2(g) + N2(g) → 2NH3(g) + 92,22 kJ
La reazione avviene dunque con cessione all'ambiente di 92,22 kJ. Scriveremo
q = − 92,22 kJ
Durante la reazione il volume del sistema si è ridotto da quello occupato dalle 4 moli dei reagenti gassosi, alle 2 moli del prodotto di reazione.
Calcoliamo allora il lavoro subito dal sistema durante la contrazione alla pressione di 1 atmosfera

Possiamo allora affermare che l'energia interna del sistema diminuisce in misura minore rispetto al calore ceduto, in quanto sul sistema viene compiuto del lavoro da parte dell'ambiente esterno pari a 4,95 kJ. Per questo motivo l'energia interna del sistema aumenterà di conseguenza e potremo scrivere
w = + 4,95 kJLa variazione complessiva dell'energia interna, tenendo conto sia del calore ceduto che del lavoro eseguito sarà allora pari a
E = q + w = ( − 92,22 kJ ) + ( + 4,95 kJ ) = − 87,37 kJ
La diminuzione di energia interna è quindi inferiore alla cessione di calore, in quanto la contrazione del sistema ha prodotto un leggero aumento di energia nel sistema.
Le reazioni che avvengono con diminuzione dell'energia interna di un sistema sono dette esoergoniche (E < 0).
Le reazioni che avvengono con aumento dell'energia interna di un sistema sono dette endoergoniche (E > 0).Entalpia (H) e termochimica
Dagli esempi visti finora sulle interazioni che avvengono tra i sistemi chimici ed il loro intorno si è potuto osservare come gli scambi energetici più consistenti (quelli che incidono in misura maggiore sulla variazione dell'energia interna del sistema) sono quelli legati allo scambio di calore q tra il sistema ed il suo intorno.
Gli scambi di energia legati al lavoro compiuto sono in genere di minor entità e sono prevalentemente legati al lavoro di espansione o di contrazione del sistema (variazioni del volume V). Ciò è dovuto al fatto che la maggior parte delle reazioni chimiche viene condotta a pressione atmosferica, lasciando che il sistema vari liberamente il proprio volume alla pressione costante di 1 atm (pressione atmosferica) in relazione al numero di moli gassose presenti all'equilibrio.Se ad esempio facciamo avvenire la reazione precedente di sintesi dell'ammoniaca in un contenitore chiuso, in cui il volume sia costante, il sistema non subisce lavoro da parte dell'ambiente.
La variazione di energia interna è la stessa (l’energia interna è infatti una funzione di stato), ma mentre in precedenza il sistema dissipava sotto forma di calore anche il lavoro di contrazione subito dall'ambiente, ora dissipa solo l'energia proveniente dalla riorganizzazione dei legami chimici.E = q + w = ( − 87,37 kJ ) + ( 0 ) = − 87,37 kJ
E' dunque fondamentale specificare se il calore di reazione (calore assorbito o ceduto durante la reazione) viene misurato a pressione costante qp o a volume costante qv.
In altre parole il calore di reazione a volume costante qv è una misura della variazione dell’energia interna del sistema (essendo w = 0)
E = qvTuttavia, come si è già detto, la maggior parte delle reazioni chimiche avviene a pressione costante, con il sistema, sottoposto alla pressione atmosferica, che risulta libero di variare il suo volume in funzione dell’eventuale variazione del numero di moli gassose che avviene durante la reazione
A questo proposito è di particolare interesse pratico l'entalpia (H), una funzione di stato il cui utilizzo risulta conveniente nel caso di reazioni isobare (a pressione costante) e poiché i processi isobari sono estremamente frequenti, il concetto di entalpia è di comune utilizzo.
L'entalpia non è una funzione di stato che discende, come l'energia interna, da considerazioni di tipo logico relativamente agli scambi energetici di un sistema, ma è una funzione di stato definita arbitrariamente e costruita secondo la seguente relazione
H = E + P.V
Consideriamo un sistema costituito da un liquido in presenza del suo vapore, contenuto in un cilindro chiuso da un pistone mobile a contatto con l’atmosfera. Se forniamo al sistema una quantità di calore qp evapora liquido, aumenta il volume e il sistema compie il lavoro w = − P (V2 –V1) sull’esterno. Quindi il primo principio può essere scritto nel modo seguente
E = E2 – E1 = qp + w = qp – P(V2 – V1)
quindi, riordinando
(E2 + PV2) – (E1 + PV1 ) = qpe dunque
H2 – H1 = H = qpSi introduce pertanto la nuova funzione di stato Entalpia H, la cui variazione H è pari al calore di reazione qp misurato a pressione costante. Ciò significa che la misura del calore di reazione effettuata a pressione costante permette di ottenere direttamente le variazioni di entalpia.
In pratica si usa misurare il calore di reazione a pressione costante e a volume costante per ottenere rispettivamente una misura di H e E. In definitiva
per reazioni a pressione costante H = qp
per reazioni a volume costante E= qv

Anche per l'entalpia non è possibile calcolare un valore assoluto, ma solo le sue variazioni (H)
Essendo una funzione di stato la variazione di entalpia non dipende dal percorso seguito durante la trasformazione, ma solo dagli stati estremi. Così potremo scrivereH = Hfinale – Hiniziale
Ciò significa che la misura del calore di reazione effettuata a pressione costante permette di ottenere direttamente le variazioni di entalpia.
Naturalmente le reazioni che cedono calore all'ambiente presentano un H negativo e sono dette esotermiche, mentre le reazioni che assorbono calore presentano un H positivo e sono dette endotermiche
reazioni esotermiche H < 0
reazioni endotermiche H > 0
Per questo motivo spesso si sente affermare che l'entalpia è il contenuto termico o calorico di un sistema (il termine stesso deriva dal greco
 = calore interno). Tale affermazione non è tuttavia corretta. Il calore non può infatti essere inteso come un'entità posseduta da un corpo (come avviene per l'energia interna o l'energia cinetica), in quanto esso è misurabile solo all'atto dello scambio tra due corpi a diversa temperatura.
= calore interno). Tale affermazione non è tuttavia corretta. Il calore non può infatti essere inteso come un'entità posseduta da un corpo (come avviene per l'energia interna o l'energia cinetica), in quanto esso è misurabile solo all'atto dello scambio tra due corpi a diversa temperatura. L'entalpia a pressione costante è una funzione di stato molto utilizzata in chimica, poiché permette di tener conto degli scambi termici che avvengono durante le reazioni (calori di reazione).
Naturalmente la variazione di entalpia associata ad una reazione (calore scambiato a pressione costante), essendo l'entalpia una funzione di stato, dipende dai valori assunti dalle variabili di stato (P, V, T).
Allo scopo di standardizzare i dati si è perciò convenuto di misurare il calore scambiato alla pressione costante di 1 atmosfera e alla temperatura di 25°C.Le variazioni di entalpia misurate in tali condizioni sono dette standard ed indicate come H°.
Inoltre, poiché non è possibile assegnare un valore assoluto all'entalpia dei diversi composti chimici in condizioni standard ( 1 atm e 25°C), si è convenuto di prendere come stato entalpico di riferimento, assegnando loro H° = 0, gli elementi chimici nel loro stato standard.In tal modo la variazione di entalpia che intercorre nella reazione di sintesi di un composto a partire dai suoi elementi costituenti può essere convenzionalmente assunta come entalpia di formazione del composto stesso.
Sia ad esempio la reazione di sintesi dell'acqua (a pressione atmosferica) a partire dagli elementi costituenti nel loro stato standard
2H2(g) + O2(g) → 2H2O(l) + 571,66 kJ
La reazione è esotermica
qp = – 571,66 kJ
ed avvenendo a pressione costante
H° = qp = – 571,66 kJ
Ma la variazione di entalpia è pari aH° = H°finale - H°iniziale = H°acqua - (H°idrogeno + H°ossigeno)
essendo convenzionalmente l'entalpia standard degli elementi pari a zero, avremo
(H° idrogeno + H° ossigeno) = 0
e quindi, l'entalpia di formazione di 2 moli di acqua sarà pari a
H° = H°acqua - (H° idrogeno + H° ossigeno) = H°acqua - 0 = H°acqua = – 571,66 kJ
Naturalmente il valore per mole sarà pari a
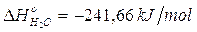
Le entalpie di formazione standard di molti composti si trovano tabulate assieme ai H° di combustione, ai H° di fusione, ai H° di evaporazione etc.
I valori di entalpia tabulati per i diversi composti possono essere utilizzati per calcolare i calori di reazione di trasformazioni chimiche anche molto complesse.
Le regole di combinazione delle entalpie dei vari composti nelle trasformazioni chimiche sono dettate dalle due leggi della termochimica: la legge di Lavoisier-Laplace e la legge di Hess
.Legge di Lavoisier-Laplace (1780)
Il calore richiesto per decomporre una sostanza è uguale al calore sviluppato durante il processo di formazione.
Possiamo riformulare in termini moderni l'enunciato di tale legge, affermando che se si inverte il verso di una reazione chimica è sufficiente invertire il segno del H, mantenendone inalterato il valore assoluto.
Prendiamo ad esempio la reazione di decomposizione dell'ammoniaca a 25°C2NH3(g) → N2(g) + 3H2(g) H° = + 92,22 kJ
Per la legge di Lavoisier-Laplace possiamo prevedere che la reazione opposta, di sintesi, presenti una variazione di entalpia uguale e contraria
N2(g) + 3H2(g) → 2NH3(g) H° = – 92,22 kJ
Legge di Hess (1840)
La somma algebrica dei calori prodotti o assorbiti durante un processo chimico a più stadi è uguale al calore prodotto o assorbito nel caso la stessa reazione avvenga attraverso uno stadio unico.
In termini moderni la legge di Hess afferma che il H di una reazione può essere ottenuto mediante somma algebrica dei H dei singoli stadi in cui si può eventualmente suddividere la reazione stessa.
Supponiamo ad esempio che la reazione
A → B
possa essere suddivisa nei seguenti stadiA → X → Y → Z → B
possiamo allora rappresentare le relative variazioni di entalpia attraverso il seguente grafico

e la legge di Hess attraverso la seguente relazione

Naturalmente entrambe le leggi della termochimica sono una diretta conseguenza del fatto che l'entalpia è una funzione di stato e i valori che essa assume negli stati iniziale e finale sono indipendenti dal percorso effettuato.Applicando le leggi della termochimica è possibile calcolare valori di H° non tabulati e calori di reazione che non possono essere misurati sperimentalmente.
Vediamo alcuni esempi
1) Vogliamo calcolare il H° di formazione del glucosio, sapendo che il suo H° di combustione è pari a - 2808 kJ/mol e che il H° di formazione dell'anidride carbonica gassosa e dell'acqua liquida sono rispettivamente -393,51 kJ/mol e -285,83 kJ/mol.
Il problema chiede di calcolare la variazione di entalpia della reazione di sintesi del glucosio a partire dagli elementi che lo costituiscono
6C + 6H2 + 3O2 → C6H12O6
Conosciamo il H° di combustione del glucosio
C6H1206 + 6O2 → 6H2O + 6CO2 H° = – 2808 kJ/mol
Per la legge di Lavoisier-Laplace il H° della reazione inversa vale
1) 6H2O + 6CO2 → C6H1206 + 6O2 H°1 = + 2808 kJ/mol
Conosciamo inoltre i H° di formazione dell'anidride carbonica e dell'acqua. Per 6 molecole possiamo scrivere
2) 6C + 6O2 → 6CO2 H°2 = – 393,51 kJ/mol . 6moli = – 2361,06 kJ
3) 6H2 + 3O2 → 6H2O H°3 = – 285,83 kJ/mol . 6moli = – 1714,98 kJ
Osserviamo ora come sommando, membro a membro, le reazioni 1), 2) e 3) si possa ottenere la reazione di formazione del glucosio dai suoi elementi, di cui vogliamo calcolare l'entalpia
6H2O + 6CO2 → C6H12O6 + 6O2 +
6C + 6O2 → 6CO2 +
6H2 + 3O2 → 6H2O =


Semplificando le specie chimiche che compaiono in entrambi i membri si ottiene
4)

Poiché dunque la reazione 4) si può ottenere come somma delle tre reazioni parziali precedenti, applicando la legge di Hess, possiamo calcolare il suo H° come somma dei tre H° parziali.
H°4 = H°1 + H°2 + H°3
H°4 = (+ 2808) + ( – 2361,06) + ( – 1714,98) = – 1268,04 kJ
2) La determinazione sperimentale del H° di formazione dell'ossido di carbonio dagli elementi è estremamente difficoltosa, in quanto, oltre all'ossido di carbonio si forma sempre una certa quantità di anidride carbonica. Il H° può essere comunque calcolato per via teorica, applicando le leggi della termochimica.
La reazione di cui si vuole calcolare il H° è la seguente
1) 2C + O2 → 2CO H°1 = ?
Tale reazione non si produce però mai da sola, poiché parte dell'ossido di carbonio reagisce con l'ossigeno per dare anidride carbonica, secondo la reazione
2) 2CO + O2 → 2CO2 H°2 = – 566,0 kJ
della quale possiamo misurare sperimentalmente il H°
3) C + O2 → CO2 H°3 = – 393,5 kJdella quale possiamo misurare sperimentalmente il H°, ottenendo la completa ossidazione, mediante combustione del carbonio con un eccesso di ossigeno.
Osserviamo ora che la reazione 3) può essere ottenuta come somma delle prime due2C + O2 → 2CO +
2CO + O2 → 2CO2 =

2C + 2O2 → 2CO2Naturalmente bisognerà tener conto che il della reazione 3) dovrà essere moltiplicato per 2 per rendere omogeneo il numero di moli con le reazioni 1) e 2).
Potremo allora scrivere
H°1 + H°2 = H°3
e quindi
H°1 = H°3 - H°2H°1 = ( – 787,0 kJ) – ( – 566,0 kJ) = – 221,0 kJ
Il H° di formazione dell'ossido di carbonio (per mole di CO) a partire dagli elementi costitutivi sarà allora pari a
H° = – 110,5 kJ/mol
3) In generale per calcolare il H° di una reazione chimica è sufficiente sottrarre alla somma delle entalpie dei prodotti, la somma delle entalpie dei reagenti.
Si voglia ad esempio calcolare il H° della seguente reazioneCaCO3(s) + 2HCl(aq) → CaCl2(s) +H2O(l) + CO2(g)
sapendo che le entalpie di formazione (valori tabulati) delle diverse specie chimiche sono
CaCO3(s) H° = – 1206,9 kJ/mol
HCl(aq) H° = – 167,2 kJ/mol
CaCl2(s) H° = – 795,8 kJ/mol
H2O(l) H° = – 285,8 kJ/mol
CO2(g) H° = – 393,5 kJ/molil H° della reazione sarà allora pari a
H° = [(– 795,8) + (– 285,8) + (– 393,5)] – [(–1206,9) + 2.( – 167,2)] = – 66,2 kJ
fonte: http://www.pianetachimica.it/didattica/documenti/Chimica_Generale.doc
Elettrochimica
Visita la nostra pagina principale
Elettrochimica
Termini d' uso e privacy
Elettrochimica